S32.2: MtDNA, microsatellites and coalescence: Tracing the colonisation of Silvereyes through the southwest Pacific
Sandie M. Degnan, Ian P. F. Owens, Sonya M. Clegg, Craig C. Moritz & Jiro Kikkawa
Department of Zoology, The University of Queensland, Brisbane, Australia, fax 61 7 3365 1655, e-mail sdegnan@zoology.uq.edu.au
Degnan, S.M., Owens, I.P.F., Clegg, S.M., Moritz, C.C., & Kikkawa, J. 1999. MtDNA, microsatellites and coalescence: Tracing the colonisation of Silvereyes through the southwest Pacific. In: Adams, N.J. & Slotow, R.H. (eds) Proc. 22 Int. Ornithol. Congr., Durban: 1881-1898. Johannesburg: BirdLife South Africa.The species complex of the widespread Silvereye Zosterops lateralis comprises nine species and 26 subspecies, including 18 taxa that have differentiated on islands. Dates for several recent colonisations are known. This system provides a unique continuum of degrees of population differentiation with which to investigate microevolutionary processes, but requires rapidly evolving molecular markers that can provide adequate resolution of the numerous recent coalescence events. Here we evaluate the adequacy of nuclear microsatellites (six loci) to reconstruct phylogenetic relationships using several different measures of genetic distance, some developed specifically for microsatellite mutation models. We show that microsatellite polymorphisms have great potential for reconstructing gene trees with greater resolution of coalescence events over time spans that may be too short for sufficient accumulation of mutations in mtDNA. All of the five genetic distance measures assessed performed well, and were able to reconstruct tree topologies that generally were in concordance with that generated by established methods using mitochondrial DNA sequences. In fact, all of the genetic distance measures had highly significant regressions on time since population separation, and a high proportion of the variance in several of the statistics could be explained by regression on time since separation. Somewhat surprisingly, the microsatellites performed well across the full range of separation times that we assessed - from no population separation through to separation over two to three million years. In spite of numerous potential caveats and even when using only a small number of microsatellite loci with widely varying mutation parameters, we conclude that microsatellite polymorphisms can provide a powerful way of assessing relationships between populations and even species.
INTRODUCTION
Over the past decade, population genetic analyses have moved from representing polymorphism as heterozygosity or number of alleles in a sample, to the more complex task of representing DNA sequence variation as a gene tree. Concomitantly, the traditional neutral evolution model of demographic history based on effective population size (Ne) has been superseded by one more appropriate to the new type of data available - the coalescent. A great advantage of coalescent models is that they can use all of the information inherent in gene trees, which in turn are much more informative than simple measures of heterozygosity or number of alleles (Harding 1996). A gene tree reconstructs the history of mutation events, and describes genealogical structure by way of providing details of the order in which pairs of lineages coalesced. Ideally, the gene tree provides sufficient information to fully resolve the ordering of coalescence events, but this ideal cannot be achieved if there are too few mutations scattered about the tree, either because the length of DNA sequenced is too short, or because coalescence events have occurred more rapidly than mutation events. Population geneticists have tended to favour mtDNA for reconstruction of gene trees, for reasons that are now well established. However, the slow mutation rate of mtDNA relative to population separation events (and thus, in reverse, coalescence events) means that there are limitations to the usefulness, particularly the resolution over short time spans, of this molecule.
This raises the question of the potential value of nuclear microsatellite polymorphisms for reconstructing gene trees with greater resolution of coalescence events over time spans that may be too short for sufficient accumulation of mutations in mtDNA. Microsatellite loci comprise numerous tandem repeats of short nucleotide sequence motifs, and are very common in the genomes of most eukaryotes (Tautz & Renz 1984). They have a substantially greater rate of mutation (average of 10-4, Weber & Wong 1993) than mtDNA, with polymorphic alleles representing an increase or a decrease in the number of repeat units. There is much still to be understood about the evolutionary processes affecting microsatellites, but already four common features have emerged from theoretical and practical studies (reviewed in Goldstein & Pollock 1997). First, mutations at microsatellite loci generally involve the addition or loss of one repeat unit, and much less frequently of several repeat units. This means that mutations tend to result in alleles with repeat numbers similar to those of the alleles from which they were derived. Thus the difference in repeat number between alleles carries information about the amount of time that has passed since they shared a common ancestral allele. Second, the mutation of repeat arrays has a complex dependence on allele size and purity of repeat pattern. Third, the mutation process is generally upwardly biased. Fourth, some selective constraints on allele size exist.
The information contained in these features is ignored by classical genetic distances based on the infinite alleles model. To overcome this limitation, several genetic distances have now been developed that are based on the, perhaps more appropriate, stepwise mutation model and that incorporate allele repeat number (for reviews, see Goldstein et al. 1995; Goldstein & Pollock 1997). In such cases, because similarity in repeat number indicates recent common ancestry, it has become possible to use this inherently phylogenetic information to generate genetic distance measures. In turn, this potentially enables microsatellite variation to be used in a way similar to some phylogenetic applications of mitochondrial DNA.
The appropriateness of microsatellite data for constructing gene trees for analysis under a coalescent model is questionable for at least two reasons. First, under a coalescent model, the information available for estimating parameters of demographic history from a gene tree is reduced if mutations are recurrent (Griffiths & Tavaré 1994). Given the rapid mutation rate of microsatellites, and the mode of evolution of microsatellite loci, the recurrence of mutations will put an upper limit on the time span of usefulness, before reversals throw a spanner in the works. Although it is clear that this time span will be shorter than that for mtDNA, it is unclear just at what point in time the limitation will arise. Second, it is important to remember that the coalescent is a population genetics model for the neutral evolution of gene trees. The neutral model provides time-scaling under the assumption of a constant molecular clock, such that there should be strict functional neutrality among the DNA variants, and the nucleotide mutation rate, which is the time-scaling factor, should be constant. The validity of this assumption for different types of molecular data can be tested only when there exists an independent time-scaling.
We present here an empirical examination of the relative usefulness of mtDNA sequence data versus microsatellite allelic data for constructing gene trees that can be analysed under a coalescent model, and with an independent time scaling. Our data are drawn from a larger study, currently underway, examining the patterns and process of phenotypic and genetic change within the species complex of the Silvereye Zosterops lateralis.
The Silvereye is a morphologically, ecologically and behaviourally variable species distributed over a large geographic range including much of the Australian mainland, with apparent repeated invasions onto islands of the southwest Pacific Ocean from eastern Australia. Mees (1969) identified a species complex comprising nine species and 26 morphological subspecies, including 18 taxa that have morphologically differentiated on islands of the southwest Pacific. Dates are known for some very recent (within the last 200 years) colonisations, can be estimated for some other recent (last several thousand years) colonisations, and are in the order of hundreds of thousands of years ago for numerous other separation events. Thus a time scale is established a priori. This system provides a valuable continuum of colonisation times and degrees of differentiation with which to investigate the partitioning and evolution of within-species variation in space and time.
Approach
For the purposes of this paper, we identify four discrete temporal categories of population separation:
Category 0: Among currently connected populations (no separation)
On the southern Great Barrier Reef, the Capricorn-Bunker (C-B) Islands are a suite of 13 coral cays that spread for approximately 100 kilometres about latitude 23o30'S (Fig.1). The islands can be geographically separated into two groups. Previous evidence from minisatellite data (Degnan 1993) and unpublished single copy nuclear data have indicated extensive gene flow both within and between these two groups.
Category 1: Among recently separated populations (1 to 200 years since separation)
We include two sets of populations in this time class. First, one cay within the Bunker Island Group described above - Lady Elliot Island (Fig. 1) - has been recolonised by Silvereyes only in the past 20 years, and shows a significant decrease in genetic variability at minisatellite loci, possibly as a result of a recent founder event (Degnan 1993). Second, historical records indicate that recent colonisations by Z. lateralis lateralis, the nominate Tasmanian race, have occurred in just the last two centuries. The Silvereye colonised the South Island of New Zealand in the 1830s, the North Island by the 1850s, and from there it crossed to Norfolk Island (Fig. 1), where it was first recorded in 1904 (Mees 1969).
Category 2: Among recently separated populations (3000 to 4000 years since separation)
Geological dating of southern Great Barrier Reef coral cays suggests that vegetation has existed on these cays for only 3000 to 4000 years (Hopley 1982). This then places an upper limit on the time of differentiation of the C-B Islands race Z. l. chlorocephala from the adjacent Australian mainland race Z. l. familiaris (Fig. 1).
Category 3: Among morphological subspecies
On the Australian mainland, several geographic populations of Silvereyes have attained subspecies status based upon morphological differentiation. We include two of these mainland subspecies in this study - Z. l. familiaris of the central east coast, and Z. l. lateralis of the offshore island state of Tasmania. A third morphological subspecies - Z. l. tephropleura - from Lord Howe Island, some 550 km off the central east Australian coast is also included (Fig. 1).
Category 4
Among discrete species. As the oldest extreme of our timescale of comparison, we include the species Z. tenuirostris from Norfolk Island, some 1200 km off the central east Australian coast (Fig. 1). Norfolk Island is of particular interest because the sympatric distribution of three species of Zosterops is considered the result of three successive colonisations, the first two having differentiated into distinct species, apparently in situ, by the time the third wave reached the island recently from New Zealand (Mees 1969). Z. tenuirostris is the second wave.
Within each of the time frames, we examine the performance of the microsatellite DNA, including how various different genetic distance measures affect that performance. Using a population phylogeny reconstructed from mtDNA as our comparison, we aim to determine the extent and accuracy of the phylogenetic information that can be recovered from the microsatellite analyses. Specifically, we use this system to address two questions: (i) can microsatellite data be successfully used to recover accurately the timescale and order of events in evolutionary history, and if so, (ii) at what time scale does the usefulness of microsatellite data expire due to accumulation of reversals?
METHODS
Sampling
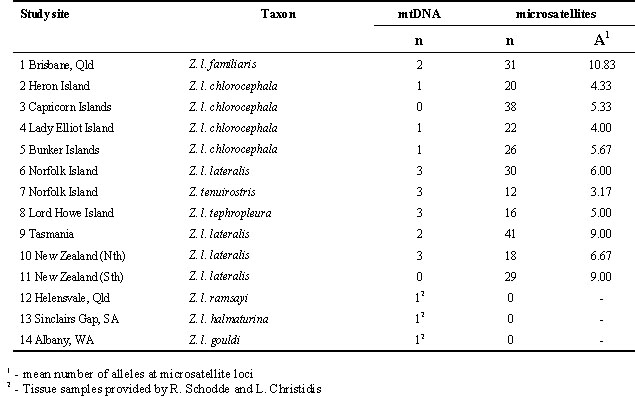
For most populations, Silvereyes were caught by trap or mist-net at each of the 14 locations shown in Fig. 1, with details given in Table 1. For each bird, total genomic DNA was extracted from a 30 to 50 m l non-destructively obtained blood sample, using a standard phenol-chloroform procedure (Sambrook et al. 1989). Morphometrics were also collected from each bird for use in associated studies not reported here. For some of the Australian mainland races, heart tissue was kindly donated by Richard Schodde and Les Christidis (see Table 1), and mtDNA was purified from these as described in Degnan & Moritz (1992).
Assaying genetic polymorphism
mtDNA
Three regions of the mitochondrial genome were amplified via the polymerase chain reaction (PCR) using two discrete sets of primers. The first set - ND6E (Edwards 1983) and CRZH2 (S. Degnan, unpublished data) - amplified part of the NADH dehydrogenase 6 (ND6) gene, the adjacent tRNAGlu and the 5' end of the control region. The second set of primers - A6TPL and COIIIHMH (E. Bermingham pers. comm.) - amplified the 3' half of the ATPase subunit 6. Amplifications were performed in 25 m l reactions containing 50 ng of total genomic DNA or 10 ng purified mtDNA, 0.4 mM of each primer, 50 mM of each dNTP, 2.5 mM MgCl2, 2.0 units of Taq DNA polymerase (Promega) and PCR buffer (10 mM Tris-HCl pH 8.3, 50 mM KCl, 0.1% Triton X-100). Amplifications were performed in an FTS-320 Thermal Sequencer (Corbett Research) using the following PCR profile: one cycle of 3 min at 94° C, followed by 6 cycles of 50 sec at 94° C, 50 s at 51° C, 45 sec at 72° C, then 25 cycles of 50 sec at 94° C, 50 sec at 55° C, 45 sec at 72° C. Amplification products were gel purified from 1.5% high melting point agarose (Promega) and used directly in ABI sequencing reactions with incorporation of flourescently labelled dNTPs. Electrophoresis and detection of sequencing reactions were performed by the University of Queensland DNA Sequencing Facility.
Microsatellites
Microsatellite variability was assayed at six loci that were isolated from a Capricorn Silvereye Z. lateralis chlorocephala, amplified and electrophoresed as described in Degnan et al. (in press). Absolute allele sizes were estimated by comparison against a known sequencing reaction and genotypes were scored by eye.
Reconstructing gene trees
mtDNA
Sequences generated as described above were compiled into concatamers and the total aligned using Clustal V (Higgins & Sharp 1989). Alignments were unambiguous and were viewed in MacClade v3.06 (Madison & Madison 1996) and subsequently saved as nexus files for further analysis in test version 4.0d64 of PAUP* written by David L Swofford. PAUP* was used to calculate Kimura two-parameter genetic distances for pair of sequences, and these subsequently were used to construct a bootstrap (1000 replicates) neighbour-joining tree. A parsimony analysis, using the branch-and-bound option for tree reconstruction with 1000 bootstrap replicates, was also performed in PAUP*. Trees generated from mtDNA sequence data were used as a basis for comparing phylogenetic reconstruction from microsatellite data, as described below.
Microsatellites
Allelic data for discrete populations were tested for conformity with Hardy-Weinberg (H-W) expectations using the Markov chain method of Guo & Thompson (1992). Five different genetic distance measures were calculated for each of 66 pairs of populations (Table 2). Variance measures were not included in the present study, but a more comprehensive analysis incorporating both Fst and Rst for comparison will be published elsewhere. The five statistics were chosen for their superior performance after a survey of published literature, including recommendations from an empirical evaluation of several distances using microsatellite data by Paetkau et al. (1997). Nei's (1972) standard distance (DS) has been widely used and acclaimed and has relatively low variance. Nei et al.'s (1983) DA has been shown, with simulated microsatellite data, to have superior performance in reconstructing phylogenetic trees (Takezaki & Nei 1996). Shriver et al. (1995) developed DSW as a modification of Nei's Dm (Nei 1973 in Takezaki & Nei 1996), incorporating the distance between each pair of alleles under consideration. Goldstein et al. (1995b) developed (d m )2, based upon the differences in mean allele size between populations. Paetkau et al.'s (1997) DLR is a genotype likelihood ratio distance. All genetic distances were estimated using a World Wide Web-based calculator that can be found at [http://www.biology.ualberta.ca/jbrzusto]. Neighbour-joining trees were constructed from the five different distance measures using the Neighbor option in PHYLIP version 3.57c (Felsenstein 1989). Tree topology was compared among the different distance measures and against the mtDNA tree. Correlation among the pairwise distances generated by the different distance statistics, and regression analyses of genetic distance against time since separation, were performed using Statview 4.0 (Abacus Concepts Inc.).
RESULTS
Relationships among populations estimated from mtDNA haplotypes
A total of 982 base pairs of mtDNA sequence was obtained for 22 individuals representing nine taxa from 14 populations (Table 1; Fig.1). The sequence comprised 226 bp of the 3' half of the ND6 gene, the adjacent tRNAGlu (68 bp), 420 bp of the 5' half of the control region, and 268 bp of ATPase 6. Nucleotide sequence data are available from the authors upon request, and a more detailed phylogenetic analysis incorporating additional Zosterops taxa will be published elsewhere. Of the 982 total characters, all given equal weight, 868 were constant, 34 were variable but uninformative, and 85 were variable and informative. Evolutionary relationships among sequences are represented by an unrooted bootstrap neighbour-joining tree (Fig. 2). Parsimony analysis generated a tree of almost identical topology (not shown).
These mtDNA trees provide the basis for assessing the relative utility of the microsatellite genetic distance measures to accurately reconstruct evolutionary relationships among populations. For comparison, then, the salient features of the mtDNA trees (Fig. 2) are:
(a) Four Z. lateralis subspecies from eight populations group into a single clade within which no relationships are resolved; that is, all sequences identified from the C-B Islands (Z. l. chlorocephala), east coast Australia (Z. l. ramsayi, Z. l. familiaris, Z. l. lateralis), New Zealand (Z. l. lateralis), and Norfolk Island (Z. l. lateralis) were almost identical, with distances not exceeding 0.2%.
(b) Following from (a), there is no resolution of known recent colonisation events, including the C-B Island's Z. l. chlorocephala from mainland Australia's Z. l. familiaris, or New Zealand and subsequently Norfolk Island by Z. l. lateralis from Tasmania.
(c) Z. l. tephropleura of Lord Howe Island forms a second monophyletic clade closely related to, but distinct from, the single unresolved clade in (a).
(d) The species Z. tenuirostris, considered to represent the second wave of colonisation of Norfolk Island from the Australian mainland, forms a distinct monophyletic clade that is substantially divergent (5.5%) from the clade containing east coast and associated island Z. lateralis subspecies. Under the assumption of a molecular clock and a commonly regarded rate of mtDNA evolution of 2% per million years, Z. tenuirostris diverged from east coast Australia Z. lateralis two to three million years ago.
Relationships among populations estimated from microsatellite alleles
Complete six-locus microsatellite genotypes (data not shown, but available from the authors upon request) were obtained for 283 individuals representing 11 geographically discrete populations (Table 1). Genotype distributions were tested for conformity to Hardy-Weinberg (H-W) expectations, as confirmation that (i) nonamplifying alleles are not present at high frequency (e.g., Pemberton et al. 1995), and/or that (ii) designated populations are not large enough to contain sufficient internal genetic structure to cause a Wahlund (1928) effect. From a total of 11 populations typed at six loci each, 63 genotype distributions could be tested; three distributions did not include two or more alleles with more than a single copy observed, so that tests could not be performed. Of 63 distributions tested, only six deviated from H-W expectations at the 5% level, and only two of these were significant at the 1% level. For each population, H-W tests were combined across the six loci. Only two populations (same as above) deviated significantly at the 5% level, and this was due to an excess of heterozygotes at locus 38 for the Capricorn Islands, and at locus 18 in Norfolk Island. This indicates generally that designated populations were not large enough to contain internal genetic structure. When H-W tests were performed for each locus across all populations, only locus 22 showed a significant deviation from H-W at the 5% level. These results indicate generally that the data set is not confounded by nonamplifying alleles.
Five different genetic distance statistics were used to estimate pairwise distances among the 11 populations for all loci combined (Table 2), and these in turn were used to generate neighbour-joining trees depicting relationships among populations (Fig. 3). One feature that is immediately apparent from comparison of the microsatellite trees against the mtDNA tree (Fig. 2) is that there is substantially more resolution in the former, although there is not complete consensus among the different distance statistics as to the exact topology of the trees. Taking the four salient features described above for the mtDNA tree, we find that, in the microsatellite trees:
(a) Populations from the C-B Islands (Z. l. chlorocephala; populations 2 to 5 in Table 1) group together as a discrete monophyletic clade in four of the five trees (except the DS tree), and always are distinct from populations of Z. l. lateralis represented by Tasmania, New Zealand and Norfolk Island.
(b) Because of this greater resolution, known recent colonisation events are revealed with some accuracy. First, in most trees, the nearest neighbour to the C-B Islands is the Brisbane population of east coast Australia. Even within the C-B Islands, Lady Elliot Island generally is the most distinct of the populations, perhaps reflecting the very recent recolonisation (and hence reduced variability due to a bottleneck effect) of this island from others within the group. The exception, again, is the DS tree. Second, the very close relationship of Z. l. lateralis populations representing the recent serial colonisation of New Zealand and Norfolk Island from Tasmania is reflected in all trees except for (d m )2. The precise topology of this Z. l. lateralis clade accurately reflects the order of colonisation events only in the DA tree, with the DLR tree also performing well.
(c) The position of Z. l. tephropleura of Lord Howe as a distinct group is supported in all but the (d m )2 tree, which oddly places Lord Howe Island inside the C-B Islands group.
(d) The species Z. tenuirostris unanimously is shown as a distinct taxon separated from Z. lateralis populations by a long branch length, reflecting the substantial divergence revealed by the mtDNA sequence data.
Time since population separation
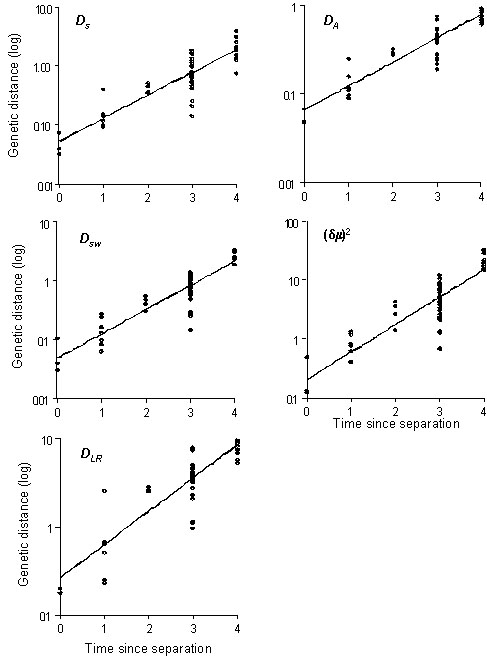
The microsatellite data used here includes 11 populations arranged along a temporal scale, ranging from no separation of populations through to ancient separation that has resulted in attainment of distinct species status. For the purposes of this paper, the temporal scale has been divided into five categories, based upon known dates of population separation for recent colonisation events through to ancient speciation events that apparently occurred some two to three million years ago (from mtDNA data described above). Measures of genetic distance (for five statistics separately) and temporal distance (according to the categories described above) were recorded for all population pairs (Table 2). Plotting the results revealed that the relationship between distance and time is best described as an exponential one (not shown), although this almost certainly is due to the non-linear time scale used in this study rather than to non-linearity of the statistics themselves.
The increase in genetic distance as a function of time was evaluated by regression, using log-transformed distance which compensated for the non-linear nature of the time scale and thus resulted in a linear relationship (Fig. 4; Table 3). It is important to note that, because each population was used in multiple data points, the assumption of independent data points is not met. Thus the results cannot be regarded as providing actual estimates of regression statistics, but rather provide a qualitative assessment of the performance of the different genetic distance measures. Given the relatively small number of microsatellite loci used in this analysis, we would expect considerable variance in measurements of genetic distance. In spite of this, upwards of 75% of the variance in DS, DA and DSW was explained by regression on time since separation. All of the distance measures, in fact, had highly significant regressions on time since separation (P<0.001). In addition to low variance and linearity, it is desirable for a genetic distance measure to have its value going to zero as the allele distributions being compared become identical. In this respect, DS, DA and DSW again perform best, because for these measures the Y-intercept predicted by the linear regression does not differ significantly from zero (Table 3). Both DLR and particularly (d m )2, however, gave values for the Y-intercept that differ from zero only at a marginal level of significance (both P<0.05).
DISCUSSION
The results from six microsatellite loci assayed in the 11 Zosterops populations confirm the power of microsatellites for studying fine-scaled population structure, and indeed for the reconstruction of gene trees for populations that in turn can be analysed under coalescent models. Over time scales that are too short for sufficient accumulation of mutations in mtDNA, the microsatellites were able to discriminate between populations separated for only decades, and could resolve relationships between populations with a fair degree of accuracy. Somewhat surprisingly, the microsatellites performed well across the full range of separation times that we assessed - from no population separation at all through to separation over two to three million years - and generally were very good at recovering phylogenetic relationships among all taxa. Although our microsatellite analyses reiterated mtDNA results of the high divergence of the distinct species Z. tenuirostris, it is difficult to comment further on the adequacy of these data for resolving interspecific relationships based upon this limited example of essentially a single pairwise comparison with Z. lateralis.
The usefulness of microsatellite data in the analyses described here depends not only upon the availability of sufficient polymorphism, but also upon the power of the distance statistics used to extract accurate phylogenetic information from the genotypes observed in each population. In this regard, the five distance measures evaluated here performed remarkably well. Because of the relatively high mutation rate of microsatellites, we would expect the genetic distance statistics to begin to plateau, and hence lose informativeness, much earlier than mtDNA. Statistics based on the infinite alleles model are expected to remain linear only over short periods of time (Goldstein et al. 1995a,b); DS, for example, theoretically is expected to remain relatively linear under the stepwise mutation model up to values of approx 0.5 (Nei 1987). On the other hand, measures such as (d m )2 and DSW were developed specifically to accommodate the pseudo-stepwise mutation of microsatellites, and so should maintain their utility beyond the other measures. In a study of bear populations, Paetkau et al. (1997) suggested that most of the measures were reaching a plateau level after less than 20,000 years of separation, and DSW and (d m )2, which may still be relatively linear even at this level, clearly lost their linearity well below the interspecific level.
Our analysis revealed no particular distance measure, or category of distance measure, that stood out as performing substantially better than any other (but see Table 4), and indeed values from each of the different measures were highly correlated (Table 3). At least part of the reason for this may be that, because many of the populations studied here are relatively small and recently colonised island populations, it is likely that genetic drift is primarily responsible for the genetic differentiation of study areas. Thus the use of accurate mutational models is not of critical importance. Because genetic drift is likely to be the primary force driving genetic differentiation in our study, the variance of the measures used may be a more important consideration than accurate mutation models. Of the measures used here, DS and DLR have relatively low variance, whereas the high variance of the other statistics, particularly (d m )2, can make it difficult to draw conclusions when using only a small number of loci.
In view of the different theoretical expectations for the performance of the different distance measures, we scored the ‘success’ of each measure in achieving several desired targets (Table 4). The scores included one point each for resolving the four elements of interest in the tree reconstructions (from Fig. 3), and one point each for achieving significance in three elements of the regression on time analysis (from Table 3), namely high r2 (variance explained by time since separation), significant F-value, and significant Y-intercept. Based upon this arbitrary ranking system, both DA and DSW score a perfect seven, but DS and DLR are close behind (Table 4). Only (d m )2 performs relatively poorly, and this probably is primarily due to the very high variance of this statistic, as discussed above.
The effect of genetic diversity on genetic distance statistics further complicates their interpretation. In particular, the magnitude of genetic distance values may be exaggerated for populations with lower diversity. Chakraborty & Nei (1976) showed that population bottlenecks can cause a marked increase in DS, and Paetkau et al. (1997) demonstrated that all of the other genetic distance measures generally will decrease towards zero as population size, and hence genetic diversity, increases. Among the population surveyed for the present study, the lowest observed genetic diversity was in the Norfolk Island population of Z. tenuirostris. All of the pairwise comparisons that fit into time category 4 involve a genetic distance between Z. tenuirostris and another population. Certainly the distances estimated for these comparisons are among the highest in this study, but this might be expected in any case based upon the high genetic divergence of this population from all others as indicated by the mtDNA data. Thus the issue of the effect of genetic diversity is confounded, but it is worth considering that the apparent linear performance of the distance measures even at this level of population separation may partly be an artefact of exaggerated distance estimates due to low genetic diversity within the Z. tenuirostris population.
It has been a common theme of recent literature that theoretical models which more accurately represent the evolutionary processes of microsatellites are needed to obtain better estimates of population differentiation measures. The widely-used stepwise mutation model may provide adequate measures for closely related populations, but has been considered too simplistic to suffice beyond a critical level of divergence and/or with the use of particular types of repeat arrays. Also, large variance in the mutation parameters among loci could be a persistent problem, such that selection of loci with similar mutation parameters might be crucial for accurate estimation of population parameters.
In spite of the numerous potential caveats, and even when using only a small number of loci with widely varying mutation parameters, we found that currently available genetic distance measures for microsatellites can suffice to provide a useful and powerful way of assessing relationships between populations and perhaps even species. The utility of microsatellite data to accurately reconstruct phylogenetic relationships of Silvereye populations over a broad range of time scales may in part be due to the modest levels of variability achieved by the microsatellite loci employed by this study. Microsatellite loci with greater rates of mutation, and especially hypervariable loci such as those that are so valuable for analyses of mating systems, may saturate too quickly to be of use beyond the level of comparison of very closely related populations. Once sufficient time has elapsed for mutation reversals to occur, phylogenetic information will be lost, so that it is important to target microsatellite loci whose observed variability is suitable for the time scale of interest. In addition, each additional locus that can be surveyed will contribute to reducing the variance of the genetic distance measures, so that total number of loci assayed should be an important consideration. With these limitations in mind, we encourage further studies to investigate more fully the precise utility of microsatellite data for phylogenetic reconstruction. We suggest that these versatile loci may make a valuable contribution to the arsenal for elucidating intraspecific evolutionary processes via the application of coalescent theory.
ACKNOWLEDGMENTS
We thank Anita Heideman for assistance in the laboratory, the Heron Island Research Station, Bernie Degnan, Andrew Hugall, Bruce Robertson and Peter Davidson for assistance in the field, Richard Schodde and Les Christidis for generously donating samples, and the National Parks and Wildlife Services of Queensland and New South Wales for permits. We greatly appreciate the efforts of John Brzustowski and David Paetkau in developing genetic distance programmes on the World Wide Web. This work was supported by grants from the Australian Research Council, National Geographic, and the Frank Chapman Fund (AMNH); we are grateful to them all.
REFERENCES
Chakraborty, R., & Nei, M. 1976. Bottleneck affects on average heterozygosity and genetic distance with the stepwise mutation model. Evolution 31: 347-356.
Degnan, S.M. 1993. The perils of single gene trees - mitochondrial versus single copy nuclear DNA variation in white-eyes (Aves: Zosteropidae). Molecular Ecology 2: 219-225.
Degnan, S.M., & Moritz, C.C. 1992. Phylogeography of mitochondrial DNA in two species of white-eye in Australia. Auk 109: 800-811.
Degnan, S.M., Robertson, B.C., Clegg, S.M. & Moritz, C.C. 1998. Microsatellite primers for studies of gene flow and mating systems in white-eyes (Zosterops). Molecular Ecology, in press.
Edwards, S.V. 1993. Mitochondrial gene genealogy and gene flow among island and mainland populations of a sedentary songbird, the grey-crowned babbler (Pomatostomus temporalis). Evolution 47: 1118-1137.
Felsenstein, J. 1993. PHYLIP - Phylogenetic Inference Package, version 3.5c. Distributed by the author, Seattle, Washington, Department of Genetics, University of Washington.
Forbes, S.H., Hogg, J.T., Buchanan, F.C., Crawford, A.M., & Allendorf, F.W. 1995. Microsatellite evolution in congeneric mammals: domestic and bighorn sheep. Molecular Biology and Evolution 12: 1106-1113.
Goldstein, D.B., & Pollock, D.D. 1997. Launching microsatellites: A review of mutation processes and methods of phylogenetic inference. Journal of Heredity 88: 335-342.
Goldstein, D.B., Ruiz-Linares, A., Cavalli-Sforza, L.L., & Feldman, M.W. 1995a. An evaluation of genetic distances for use with microsatellite loci. Genetics 139: 463-471.
Goldstein, D. B., Ruiz-Linares, A., Cavalli-Sforza, L. L., & Feldman, M. W. 1995b. Genetic absolute dating based on microsatellites and the origin of modern humans. Proceedings of the National Academy of Sciences, USA 92: 6723-6727.
Griffiths, R.C., & Tavare, S. 1994. Simulating probability distributions in the coalescent. Theoretical Population Biology 46: 131-159.
Guo, S.W., & Thompson, E.A. 1992. Performing the exact test of Hardy-Weinberg proportion for multiple alleles. Biometrics 48: 361-372.
Harding, R.M. 1996. New phylogenies: an introductory look at the coalescent. In: Harvey, P. H., Brown, A. J. L., Smith, J. M., & Nee, S. (eds) New Uses for New Phylogenies; Oxford, Oxford University Press: 15-22.
Higgins, D.G., & Sharp, P.M. 1989. Fast and sensitive multiple sequence alignments on a microcomputer. Cabios 5: 151-153.
Hopley, D. 1982. The geomorphology of the Great Barrier Reef: Quaternary development of coral reefs. New York, Wiley.
Madison, W.P., & Madison, D.R. 1992. MacClade: analysis of phylogeny and character evolution, version 3. Sunderland, Massachusetts, Sinauer Associates.
Mees, G.F. 1969. A systematic review of the Indo-Australian Zosteropidae (Part III). Zoologische Verhandelingen, Leiden 102: 1-390.
Nei, M. 1972. Genetic distance between populations. American Naturalist 106: 283-292.
Nei, M. 1987. Molecular and Evolutionary Genetics. New York, Columbia University Press.
Nei, M., Tajima, F., & Tateno, Y. 1983. Accuracy of estimated phylogenetic trees from molecular data. Journal of Molecular Evolution 19: 153-170.
Paetkau, D., Waits, L.P., Clarkson, P.L., Craighead, L., & Strobeck, C. 1997. An empirical evaluation of genetic distance statistics using microsatellite data from bear (Ursidae) populations. Genetics 147: 1943-1957.
Pemberton, J.M., Slate, J., Bancroft, D.R., & Barret, J.A. 1995. Nonamplifying alleles at microsatellite loci: a caution for parentage and population studies. Molecular Ecology 4: 249-252.
Sambrook, J., Fritsch, E.F., & Maniatis, T. 1989. Molecular Cloning. A Laboratory Manual, 2nd ed. Cold Spring Harbor, New York, Cold Spring Harbor Laboratory.
Shriver, M.D., Jin, L., Boerwinkle, E., Deka, R., & Ferrell, R.E. 1995. A novel measure of genetic distance for highly polymorphic tandem repeat loci. Molecular Biology and Evolution 12: 914-920.
Slatkin, M. 1995. A measure of population subdivision based on microsatellite allele frequencies. Genetics 139: 457-462.
Takezaki, N., & Nei, M. 1996. Genetic distances and reconstruction of phylogenetic trees from microsatellite DNA. Genetics 144: 389-399.
Tautz, D., & Renz, M. 1984. Simple sequences are ubiquitous components of eukaryotic genomes. Nucleic Acids Research 12: 4127-4138.
Wahlund, S. 1928. Zusammensetzung von populationen und korrelationserscheinungen vom standpunkt der vererbungslehre aus betrachtet. Hereditas 11: 65-106.
Weber, J.L., & Wong, C. 1993. Mutation of human short tandem repeats. Human Molecular Genetics 2: 1123-1128.
Table 1. Details of Silvereye populations represented in mtDNA and microsatellite analyses. Populations 2 to 5 represent the Capricorn-Bunker Islands.
Table 2. Genetic distances estimated from microsatellite data and used to generate neighbour-joining trees shown in Fig. 3. Population identification numbers follow those in Table 1. Categories of ‘time since separation,’ used to generate Figs. 4 & 5 are shown for each pairwise comparison.
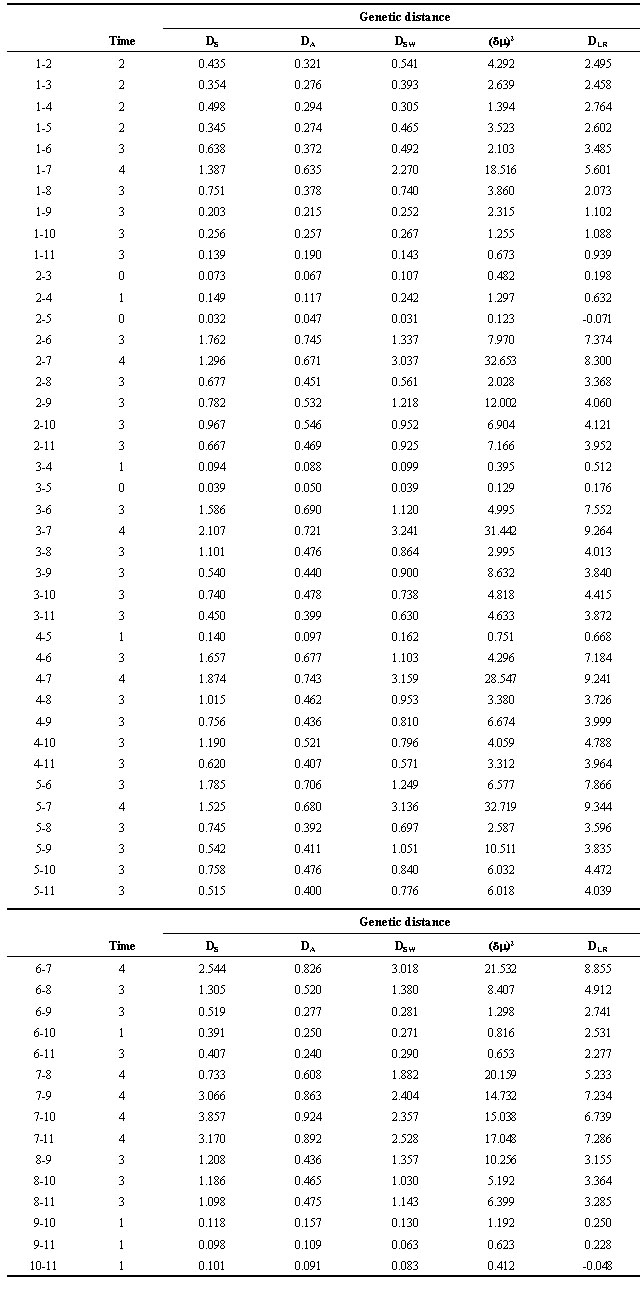
Table 3. Regression statistics for Fig. 4.
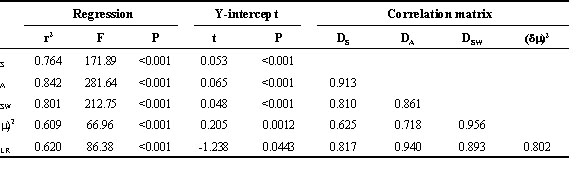
Table 4. Performance scores of five genetic distance statistics used in this study.
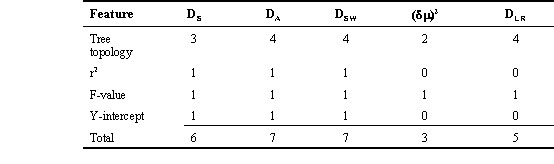
Fig. 1. Location of 13 Zosterops lateralis (Silvereye; 1-6, 8-14) and one Z. tenuirostris (7) populations analysed for mtDNA and/or microsatellite polymorphism. (A) Australia and the southwest Pacific islands showing location of inset. Solid line within the Australian mainland indicates the inland limit of distribution of the Silvereye. (B) Inset details the Capricorn-Bunker Islands on the southern Great Barrier Reef; open circles show islands not included in the present study.
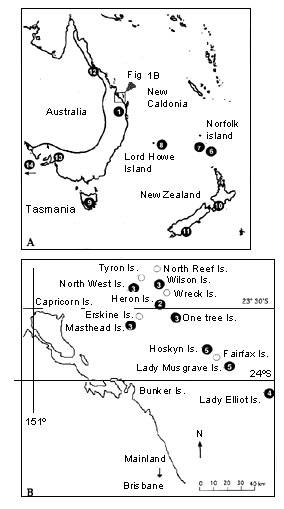
Fig. 2. Unrooted neighbour-joining tree depicting relationships between mtDNA haplotypes identified from 12 Z. lateralis and Z. tenuirostris populations. Numbers above branches represent branch lengths, and those below are group frequencies from 100 bootstrap replicates. Numbers in parentheses beside taxonomic names are the number of identical sequences represented. The tree was generated from Kimura 2-parameter pairwise distances.
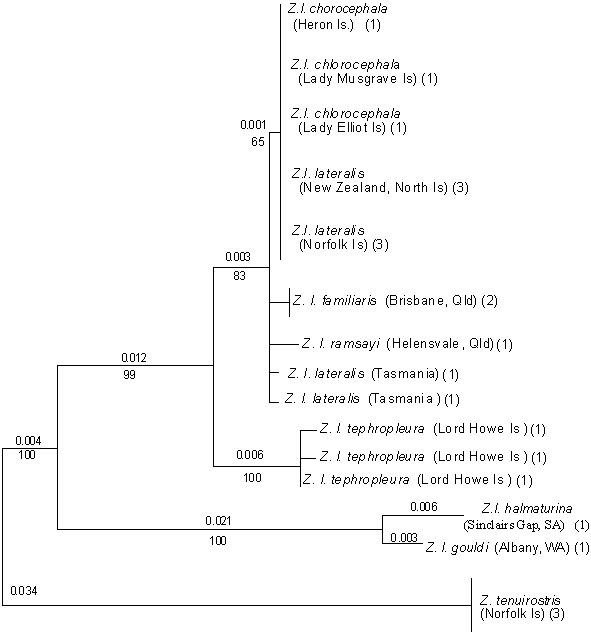
Fig. 3. Unrooted neighbour-joining trees showing relationships between microsatellite genotypes identified from ten Z. lateralis and one Z. tenuirostris populations. Each tree was generated from a different genetic distance statistic, as indicated.
Fig. 4. Plot of genetic distance (log scale) against temporal distance for all pairs of populations analysed for microsatellites. Each of five different genetic distance statistics used is shown separately, with regression lines indicated. The axis ‘Time since separation’ corresponds to categories described in the ‘Approach’ section of this paper.