S13.5: The implications of a priori versus a posteriori designation of analytical methods
Scott M. Lanyon1 & Kevin E. Omland1,2
1James Ford Bell Museum of Natural History, University of Minnesota, 1987 Upper Buford Circle, St. Paul, Minnesota 55108-6097, USA, e-mail slanyon@biosci.umn.ed; 2komland@biosci.umn.edu
Lanyon, S. M. & Omland, K. E. 1999. The implications of a priori versus a posteriori designation of analytical methods. In: Adams, N.J. & Slotow, R.H. (eds) Proc. 22 Int. Ornithol. Congr., Durban: 762-768. Johannesburg: BirdLife South Africa.The availability of a diversity of algorithms and options for analysing molecular data has been greeted with enthusiasm by avian systematists but has led to an important, but relatively unrecognised, problem in systematics. Systematists have partially abandoned the scientific method. Rather than designating a priori the analytical approach to be used, reporting the results of that approach, and then further examining the data with additional analyses selected a posteriori, many investigators adopt the a posteriori approach alone (although this is rarely the way it is written in the methods sections of publications). The importance of designating, and implementing, an analytical approach that is selected a priori is discussed. We argue that two a priori approaches must be designated to produce the two distinct products of systematic study: a well-resolved estimate of phylogeny (e.g., a single fully dichotomous cladogram) and a well-supported estimate of phylogeny (e.g., one that includes only reliable nodes as indicated by indices of support). In addition, we discuss the importance of also conducting a posteriori analyses to further explore patterns within the data.
INTRODUCTION
One of the great advances in systematics in the latter half of this century was a major shift in systematic philosophy that led to the development and use of rigorous methods. This revolution was accompanied by a more pervasive revolution in the sciences and that is the availability of cheap computing cycles on many investigators’ desk tops. However, in this flurry of advances, systematics has failed to recognise two problems that we believe to be pervasive, and potentially quite damaging, to our science. The first is the abandonment, or at least the weakening, of the scientific method and the second is confusion regarding what it is we are trying to produce.
The appropriate place to start this discussion is with a review of the Scientific Method. Webster’s Dictionary defines the Scientific Method as ‘Principles and procedures for the systematic pursuit of knowledge involving the recognition and formulation of a problem, the collection of data through observation and experiment, and the formulation and testing of hypotheses.’ Simply put, we state our question, detail our methodology, collect our data, describe our results, and offer an interpretation of our findings. Scientists conduct their research and prepare their reports following this basic outline. Because the results of one study (one iteration of the scientific method) inform subsequent studies, we can think of SCIENCE as being a endless repetition of the cycle of the scientific method.
Two primary advantages come from the rigorous application of the scientific method. First, the scientific method insures clarity in describing the details of a study. Historically, systematists failed to state their methods adequately such that independent researchers could not repeat their results. Advances in analytical methods, including increased rigor in how characters are identified and described, have improved the repeatability of systematic biology dramatically. The second benefit of employing the scientific method is the limitation of the impact of investigator bias on science.
The recognition of the potential for investigator bias is an important part of any scientific study. Sources of bias may be divided into three classes as a function of when they are introduced into a study. Two of these classes are well known, and it is the third, less well known class, that is the subject of this paper. A priori biases are those that are introduced during the initial design of the study. In systematics, such biases can and do influence choice of ingroup taxa, outgroup taxa, characters, analytical model, and criteria for identifying the ‘best’ tree. Although a priori biases can seriously affect the outcome of scientific studies, explicit discussion of how studies are designed allows others to look for and to detect such biases.
A posteriori biases are those that are introduced during the interpretation of the results of the study. This is a less subtle form of bias and most scientists are trained to look for, and to avoid such biases. For example, an investigator may ‘know’ from previous studies, that a group is monophyletic and, therefore, may conclude that a finding of polyphyly in the present study, is incorrect and represents misleading patterns in the current data set. Once again, explicit discussion of how this conclusion was reached allows others to look for and to detect biases in the interpretation of the results.
Current systematic studies have introduced a third and potentially significant source of bias that systematists do not document, and of which they are largely unaware. We name this third class of biases ‘course of analysis’ or a mediori biases, literally biases ‘from the middle.’
COURSE OF ANALYSIS BIASES
When systematic studies of DNA sequence data are reported, the manuscripts reflect the scientific method, but it is rarely a true reflection of the study that was performed. The ease with which we can now conduct sophisticated analyses of our data, has introduced a new wrinkle into our science.
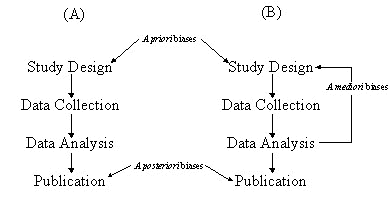
Discussions with avian molecular systematists, ourselves included, suggest that there are very few systematic studies published today that did not involve multiple ‘preliminary’, and unreported, analyses of the data before all the data was in hand. Rather than a simple execution of the sequential steps of the scientific method as indicated in our publications, systematic studies are in reality, many iterations of the first three steps of the scientific method before the publication emerges (Fig. 1).
An extreme example of this kind of bias is given by Theriot et al. (1995). In describing the methods used in their study of phylogenetic affinities of Barney, the authors state ‘We added or discarded characters until we achieved the results we believed, then stopped.’ Multiple analyses were performed and these results were used to modify the study design until the desired answer was obtained. While this is an extreme example, we contend that less blatant ‘course of analysis’ biases are rampant in systematic biology.
Consider the following hypothetical example. A study is designed to examine 25 ingroup taxa and two outgroup taxa. The analytical method selected is a parsimony analysis using equal weights for all characters and all character-state changes. The source of characters is the mitochondrial cytochrome-b gene. The investigator begins the study and when approximately 25% of the data has been collected, a preliminary parsimony analysis is conducted. Having recently read an article recommending 2X weighting of transversions over transitions, this analysis too is performed. The results are very encouraging, especially from the 2X weighting. However, there is some indication that long-branch attraction may be causing difficulty in one area of the topology. The investigator chooses to add three more ingroup taxa for which material has become available.
With 50% of the data in hand, the investigator conducts another set of analyses: 2X weighting, weighting by codon position, and a maximum likelihood analysis. The results remain promising and both codon position and maximum likelihood seem to provide better resolution. Rooting of the ingroup seems to be a problem, however, and five additional outgroup taxa are added.
With 75% of the data collected the investigator fears that resolution of the root will be difficult for two reasons. One outgroup taxon seems to be extremely homoplasious and cytochrome-b, although informative about many events, seems relatively uninformative about deeper nodes. The decision is made to drop the suspect outgroup taxon and to sequence a portion of the ND2 gene.
Finally, all the remaining data are collected and a maximum likelihood analysis is performed. The paper that results from this study describes the 28 ingroup taxa, six outgroup taxa, the use of maximum likelihood, and the selection of cytochrome-b and ND2 as sources of systematic characters. The paper does not report the initial study design, nor does it describe the process by which this initial design was modified to achieve the final design. Instead of a single systematic study, as is ultimately reported, the hypothetical study we’ve described was in fact four systematic studies. We believe that systematic studies that take years to complete and which involve many tens of taxa, are more likely a series of hundreds of systematic studies, only one of which (the last) is reported.
While it is true that this last study is well reported and can be replicated, what is not documented are the many many opportunities for the introduction of ‘course of analysis’ biases into this research. Why did the investigator suspect long-branch attraction and add more ingroup taxa? Because the preliminary results were not consistent with his/her expectations. Why did the investigator use maximum likelihood rather than the equal weight parsimony analysis originally selected? Because maximum likelihood provided better resolution. All of these decisions should be, but are not, documented in modern systematic studies.
We believe that a mediori biases are particularly problematic for biochemical systematic studies. As implied in the above example, there exists a plethora of analytical methods for the analysis of sequence data. New methods, and new modifications of existing methods, are developed at a pace that has prevented the development of any consensus about the ‘best’ analytical approach. Consequently, there is a tendency, indeed there is strong pressure, to modify our methodologies during the course of our research.
WHAT ARE THE GOALS OF SYSTEMATIC STUDIES?
A second factor that makes a mediori biases an especially thorny problem in systematics, is the confusion that exists within systematic biology concerning the nature of the goals of our research. Are we trying to produce the best summary of the pattern among terminals given a particular analytical approach and suite of assumptions, or are we trying to produce the best estimate of evolutionary history? If we are only interested in the former, then the impact of biases, of any kind, are relatively unimportant. Even if we conduct a thousand different analyses before deciding on a single one to report, this does not affect our conclusion that we have found the best summary of the data given our analytical methods and assumptions.
Minimising a mediori biases is critically important, however, if we are also interested in producing the best estimate of evolutionary relationships of our study organisms. A thousand different analyses conducted over the course of a project provides ample opportunity for selecting, in a biased fashion, from among the myriad possible combinations of ingroup taxa, outgroup taxa, algorithm, selection criterion, and character suites. In the hypothetical example given above, the investigator made decisions to modify the study design because the results of preliminary analyses were unsatisfying (i.e., they did not fit expectations). This is a clear example of bias which, due to incomplete documentation, is hidden from the scientific community and cannot be assessed.
It is important to note that it isn’t just the selection of the methods for deriving a ‘best’ topology that is subject to a mediori biases. Systematists are also selecting methods for evaluating nodal support in the ‘best’ topology in an iterative, and undocumented, manner. As was true for the derivation of the shortest topology, there are multiple ways of assessing the strength of support for nodes (e.g., bootstrapping characters [Felsenstein, 1985]; jackknifing taxa [Lanyon, 1985]; and decay indices [Bremmer, 1988]). Over the course of the study there is ample opportunity to make choices regarding the approach to be used as well as whether or not a threshold value will be specified and what its magnitude will be.
SOLUTIONS
How then can we modify our systematic studies to control a mediori biases? There are really just two ways to answer this question. We can we can bring these potential sources of bias out into the light or we can eliminate them from our studies. Bringing these biases into the light can be accomplished by a thorough documentation of the methods actually used over the course of the entire study. This has the advantage that it enables investigators to take advantage of methodological advances that occur after initial project design. The primary disadvantage is that methods sections for systematic papers may well become larger than the rest of the paper put together.
An alternative is to do our best to eliminate a mediori sources of bias. Explicit use of the scientific method will accomplish this goal. By specifying the study design a priori, both tree generation methods and tree assessment methods, and employing that design without alteration, this source of bias can be eliminated. Note that use of the scientific method still enables investigators to employ a variety of methods specified a posteriori to help them further interpret their results. However, the a posteriori nature of these additional analyses, and associated biases, is specified clearly.
EXAMPLES
We regret to say that we know from personal experience how real a mediori biases are, and how difficult it can be to eliminate them from a scientific study. Our concurrent molecular systematic study of New World blackbirds provided us with the opportunity to test our ideas of controlling this class of biases in our research. Convinced, as we were, of the need to avoid ‘course of analysis’ biases, we were astonished at the number of times we found ourselves struggling under this self-imposed constraint. We found that the temptation to abandon our a priori approach was very strong.
In this section, we use examples from our own work to illustrate the different ways to address a mediori biases in systematic biology. This seemed to be the best way to avoid pointing out actual or potential biases perceived in the work of our colleagues.
We feel that, in most cases, the best solution is to specify study design a priori and to resist altering that design. As an example, in a study of the evolution of inter-specific brood parasitism, Lanyon (1992) examined 852 base pairs from the cytochrome-b gene for twenty-six blackbird taxa using a parsimony analysis with all character-state changes given equal weights. This study design was selected a priori, was not altered during the course of the study, was implemented, and the results were reported. In this instance, sticking to the a priori design was very easy. First, this particular study was tangential to a larger study on icterid systematics (Lanyon and Omland, submitted) and was initiated once all the data had been collected. Therefore, there was little opportunity for ‘course of analysis’ feedback on study design. Consequently, there were few ‘preliminary analyses’ conducted. Secondly, because the design produced a well-resolved, fully dichotomous topology, there was no ‘incentive’ to alter the design.
In contrast, in our study of Icterus phylogenetic relationships (Omland, Lanyon and Fritz, submitted) we experienced more difficulty in following our prescription of choosing analysis methods a priori. Here, we use this example to illustrate three points about the methods and challenges of implementing this a priori approach.
First, choosing an analysis strategy a priori does not require one to specify all the detailed mechanics of the analysis at the outset. For example, we chose to use six parameter weighting (following Williams and Fitch, 1989) as our primary a priori weighting method because it accounts for both transition-transversion biases, as well as base composition biases. However, we specified a priori only the procedure we would use to obtain the six-parameter weights, not the weights themselves. We said that we would obtain a shortest tree based on equally weighted parsimony and then reconstruct changes onto that tree to obtain the frequency of each of the six substitution classes (A-C, A-G, A-T, C-G, C-T, G-T). We chose this strategy at the outset of the study, but obviously did not actually obtain the six-parameter weights until we had a complete data set and an equally weighted tree.
Second, we also chose a priori two alternative weighting schemes for comparison with the six-parameter weighting to help us evaluate the degree to which our results were dependent on algorithm choice. We chose equally weighted parsimony, and parsimony with transversions given three times the weight of transversions.
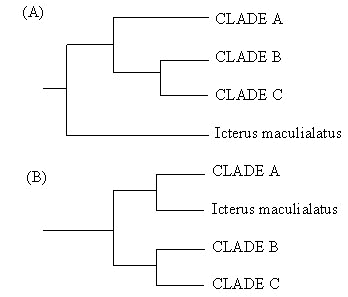
The heuristic search results obtained from the three weighting schemes differed somewhat in the placement of the three main clades of orioles (labelled A, B and C), and one problematic taxon, Icterus maculialatus (Fig. 2.). Six-parameter weighting found a single shortest tree identifying maculialatus as sister to all other orioles. In contrast, equal and 3x weighting both produced three most parsimonious trees that all placed maculialatus sister to clade A. Independent analysis of each single gene data set by all weighting schemes also supported the equal/3x tree. The only data set/weighting combination that produced evidence that maculialatus is sister to all other Icterus was the six-parameter analysis of the combined data (our a priori designated analytical approach). However, even that search produced a second island of trees just 0.03% longer, that matched the equal/3x trees.
Therefore, our a priori study design produced a weakly supported topology that conflicted with the topology found by all other analyses we conducted. Having reported this outcome, we went on to conclude that the 6-parameter analysis was uninformative about placement of maculialatus (based on the existence of two topological islands of nearly identical tree length). Instead, we selected the equal and 3x-weighted tree as our single best estimate of the phylogeny, and we plan to use it to reconstruct the history of plumage evolution.
Finally, the Icterus example makes it clear that following this approach can be challenging even for those committed to its importance. It is tempting to simply not report the results of the six-parameter searches because these results in some ways complicate our conclusions. It is even tempting to only report the results of the equally weighted searches for simplicity; the results of the 3x searches were so similar. However, we feel that it is crucial to report all of the steps we have taken in this research so that others can fully interpret, evaluate, and perhaps repeat our study. Indeed, the difficulty we encountered in implementing this philosophy is perhaps the strongest evidence of the many subtle ways that ‘course of analysis’ bias influences our science.
Although we believe that a priori designation of methods is ideal, we recognise that it is not always possible. Sometimes, there are good reasons to allow the study design to be produced in an agglomerative fashion. For example, in a study of Toucan phylogenetic relationships, Barker and Lanyon (unpub ms.) began with relatively few analytical approaches but ended up conducting many different analyses of the complete dataset. However, unlike the hypothetical example given earlier, once an analytical approach was employed it became a part of the study design. Because we wished to minimise a mediori biases that we knew could be introduced with this study design, we used the phylogenetic framework approach (Lanyon 1993) to summarise all the results. Having first identified the strongly supported nodes resulting from each analysis, the authors were able to produce a single phylogenetic framework that summarised all analyses.
CONCLUSIONS
The scientific method is a wonderfully powerful tool. Unfortunately, over the past twenty years, systematic biology has slowly, and unknowingly, abandoned this method.
Before desktop computers became available, we collected all our data and set up for one analysis, did the analysis, reported the results, and maybe did some partial analyses to explore these results. Now we are often analysing partial data as it is accumulated and using a variety of methods with a full intermixing, and unspecified interdependence of, observation and method selection. The consequence has been the loss of a meaningful distinction between methods selected a priori vs. those developed as the project progressed. We are certain that a mediori biases, introduced as a result of a dangerous cycle of preliminary analyses and study design modification, are rampant in systematic studies. To minimize the introduction of a mediori biases into systematic studies, we advocate for the specification of methods a priori and the implementation of those methods without alteration.
REFERENCES
Bremer, K. 1988. The limits of amino acid sequence data in angiosperm phylogenetic reconstruction. Evolution. 42: 795-803.
Felsenstein, J. 1985. Confidence limits on phylogenies: an approach using the bootstrap. Evolution. 39: 783-791.
Hillis, D. M. 1998. Taxonomic sampling, phylogenetic accuracy, and investigator bias. Systematic Biology 47: 3-8.
Lanyon, S. M. 1985. Detecting internal inconsistencies in distance data. Systematic Zoology 34: 397-403.
Lanyon, S. M. 1992. Interspecific brood parasitism in blackbirds (Icterinae): A phylogenetic perspective. Science 255: 77-79.
Lanyon, S. M. 1993. Phylogenetic frameworks: Towards a firmer foundation for the comparative approach. Biological. Journal of the Linnaean Society. 49: 45-61.
Theriot, E. C., Bogan, A. E. & Spamer, E. E. 1995. The taxonomy of Barney: Evidence of convergence in hominid evolution. Annals of Improbable Research 1: 3-7.
Fig. 1. Sources of bias in (A) the scientific method and (B) the systematic method.
Fig. 2. Alternative topologies of relationships within the genus Icterus resulting from (A) an a priori 6-parameter weighting anaylsis and (B) various a posteriori analyses described in the text.