S13.4: The effect of structure and function of a protein on evolution in protein-coding genes: Problems in retrieving phylogenetic signal
Carole S. Griffiths
1Department of Ornithology, American Museum of Natural History, Central Park West at 79th Street, New York 10024, USA, fax 1 212 769 5759, e-mail csg@amnh.orgGriffiths, C. S. 1999. The effect of structure and function of a protein on evolution in protein-coding genes: Problems in retrieving phylogenetic signal. In: Adams, N.J. & Slotow, R.H. (eds) Proc. 22 Int. Ornithol. Congr., Durban: 754-761. Johannesburg: BirdLife South Africa.
Homoplasy can cause problems in retrieving phylogenetic signal in protein-coding genes. One solution is to give less weight in phylogenetic analysis to unreliable characters, i.e., those that evolve too rapidly for the level of the analysis. Identifying rapidly evolving sites is not straightforward. Rates of change vary among sites in sequences and this variation can be confounded by different rates of synonymous versus non-synonymous substitutions and differences in rates of transversions and transitions. This paper reviews the methods used to identify rapidly evolving sites in protein-coding genes. A new direction to this research is then discussed, one that incorporates information about the structure and function of the protein.
INTRODUCTION
The initial use of molecular data in systematics brought with it the hope that these data would answer the difficult questions that could not be solved using morphological data. It also brought the expectation that these data would not be subject to the same problems of convergence as morphological data (Sibley and Ahlquist 1987). However, problems of convergence in sequence data were recognised quickly (Brown 1979), and have become more apparent as molecular research increases (Irwin et al. 1991; Graybeal 1993; Helm-Bychowski & Cracraft 1993; Hackett 1996). These problems are caused by multiple substitutions at sites in sequences. When changes occur several times along a branch in a phylogenetic tree, accurately estimating the ancestral character state at the node leading to that branch becomes problematic (Kim 1996). Multiple substitutions may not cause problems if taxa are closely related. However, as divergence times among taxa increase, the number of sites differing among these taxa also increases and it becomes more probable for a new mutation to occur at a site that already varies than at an invariant site (Moritz et al. 1987). Recovering phylogenetic signal then becomes difficult.
One solution offered to the problem of homoplasy in a set of sequences is to add more data (Brower and DeSalle 1994). This assumes that homoplasy is randomly distributed among sites; as more data are added, the random error caused by homoplasy will be outweighed by the phylogenetic signal in the data (Brower and DeSalle 1994). Although some researchers have obtained more reliable hypotheses by combining gene sequences (e.g., Olmstead and Sweere 1994; Sullivan 1996), others have not found this supposition to be supported. For example, a study using all mitochondrial protein-coding genes for 19 taxa from all major animal lineages demonstrated that simply adding more data was not effective in retrieving phylogenetic signal (Naylor and Brown 1997).
An alternative solution is to give less weight in phylogenetic analysis to unreliable characters, i.e., those that evolve too rapidly for the level of the analysis (Swofford et al. 1996). This can be accomplished either by differentially weighting these characters in parsimony analysis or by incorporating information about rates into a maximum likelihood model or distance analysis. However, identifying these rapidly evolving sites is not straightforward. Rates of change vary among sites in sequences and this variation can be confounded by different rates of synonymous versus non-synonymous substitutions and differences in rates of transversions and transitions.
Approaches to identifying rapidly evolving sites of protein-coding genes have changed as DNA sequence data have become more prevalent and information about patterns of change has become more available. This paper will briefly review the changes in these approaches and then discuss a new direction for this research.
REVIEW
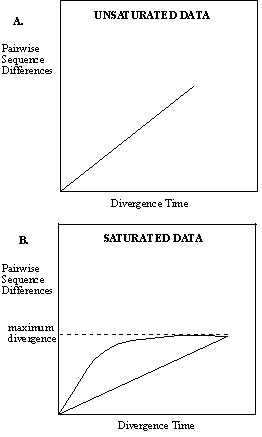
All methods of phylogenetic inference have a model of sequence evolution, either implicit or explicitly stated, that serves as a framework for analysing mutations. In the initial efforts to analyse DNA sequence data, the model viewed sequence characters as part of an independent, linear system in which changes occurred randomly and independently, with an equal probability of mutation among sites. One method used to assess the potential problem of multiple hits in these data was to determine if substitutions were saturated. This was done graphically by plotting sequence differences between pairs of taxa against an estimate of time of divergence of the taxa (Brown 1983; Moritz et al. 1987). For unsaturated data, as time since divergence of taxa increases, pairwise sequence differences between taxa also increase and the graphical display of these differences shows a monotonic increase (Fig. 1A). For saturated sites, as time of divergence between taxa increases, pairwise differences in sequences reach a maximum. The graphical display of this phenomenon is a curve which asymptotes (Fig. 1B). Using this approach, initial analysis of total sequence divergence within mammals indicated that saturation of substitutions occurred between species whose time of divergence was 30 million years ago (Brown et al. 1979).
As sequence data accumulated, insights into the patterns of evolution provided enhancements to the model. One of the first insights was that transitions occur at a higher rate than transversions in the mitochondrial genome (Brown et al. 1982). Sequence data could then be partitioned into subsets of transitions and transversions for separate saturation analyses. Saturation of transition substitutions could be corrected through the use of transversion parsimony (Helm-Bychowski and Cracraft 1993) or through differentially weighting transitions and transversions (e.g., Friesen and Anderson 1997).
The second major pattern of evolution revealed in protein-coding genes was that rates of change varied among the three codon positions (Brown 1985). In particular, substitutions at third codon positions, primarily synonymous substitutions, were the most rapidly-occurring. A second partitioning strategy could be performed by dividing data into three subsets representing each codon position. For phylogenetic inference, differences in rates could be accommodated through differential weighting of codon positions (Edwards et al. 1991) or by filtering saturated third codon positions (Edwards et al. 1991; Kornegay et al. 1993).
Combining these two patterns engendered a further enhancement to the model of sequence evolution. A more detailed analysis of saturation could be performed by dividing sequences into six subsets, transitions and transversions at each codon position. Empirical results of these analyses generally demonstrated saturation in transitions at third positions (e.g., Hackett 1996), and sometimes in first positions. Recognising these saturated partitions of data, and downweighting them, improved the results of phylogenetic analysis at higher systematic levels (Yoder et al. 1996).
This seemed to be the limit to the logical partitioning of sequences. However, in protein-coding genes not all changes at all locations are equally likely. For example, cytochrome b is one of the more conservatively changing mitochondrial genes because many positions are invariant over vertebrates, apparently reflecting constraints based on protein structure (Howell 1989; Degli Esposti et al. 1993). This relationship between the function of the protein product and evolution of the gene is incorporated into the final extension of the saturation method.
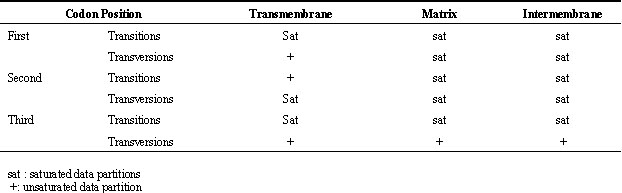
The cytochrome b protein spans three regions of the mitochondrion (the inner matrix, the inner membrane, and the outer, intermembrane area; Howell, 1989; Degli Esposti et al., 1993). If cytochrome b sequences are divided into subsets corresponding to these three domains, transitions and transversions at each position within each domain can be graphed yielding a total of 18 plots. In an analysis of the Falconidae, this resulted in 13 saturated partitions (Table 1; Griffiths 1997). When these saturated partitions were differentially weighted, phylogenetic inference was improved; the hypotheses derived from molecular and morphological data were congruent.
These insights changed the initial model of evolution of DNA sequences by recognising variation in rates of evolution among sites. However, changes at sites were still viewed linearly and assumed to be independent. There are limitations using this approach. Partitioning data, and a limit on the number of variable sites within certain partitions, can lead to very small partitions. The small number of differences may inflate the effect of sampling error (Swofford et al. 1996).
More substantive criticisms are that some amino acid changes require multiple nucleotide mutations, and that selection at the amino acid level may effect evolution of the gene coding for that protein. Thus, a major assumption of the model is violated; changes at sites may not vary independently (Swofford et al. 1996).
CURRENT DIRECTION OF RESEARCH
To identify rapidly changing sites in protein-coding genes requires changing the model of sequence evolution from a linear model to one considering sequence variation as part of a complex, hierarchical system. This model more accurately accommodates the data by characterising patterns and rates of sequence evolution with phenotypic features, thus incorporating knowledge of the structure and function of proteins into phylogenetic inference (Goldman et al. 1998). However, characterising nucleotide changes that may be correlated with structural constraints at the protein level is difficult. To ease the analysis, variation in protein-coding genes can be investigated at the amino acid level in translations of sequences.
This is currently being accomplished through two different approaches. The first derives site specific probabilities of change between amino acids by evaluating amino acids at each residue location in translations of gene sequences. These probabilities are then incorporated into models for phylogenetic inference. Koshi and Goldstein (1998) use fitness functions of each amino acid, based on chemical and physical properties of the amino acids at each site, to derive mutation matrices. Goldman et al. (1998) derive amino acid transformation probabilities for different structural categories and then fit each site into one of these categories.
The second approach is tree-based. Well-accepted phylogenetic hypotheses are used to find patterns of change at the amino acid level that are related to structure and functions, to determine differences in rates among residues. In an analysis of exemplar taxa using sequences of the entire mitochondrial genome, a known phylogeny was inferred from a small subset of sites coding for structurally important amino acids (Naylor and Brown 1997).
Griffiths (1998) investigated molecular evolution and phylogenetics of the mitochondrial protein-coding gene to provide a more detailed analysis of patterns of change. Cytochrome oxidase III was used because the structure of this protein had been elucidated through X-ray crystallography (Tsukihara et al. 1996). In this investigation, rapidly evolving amino acid residues (labile residues) were identified. Phylogenetic tests were then run to test the effect of downweighting the labile codons (corresponding to the labile residues) on the accuracy of phylogenetic inference. These tests attempted to retrieve a known phylogeny of 16 vertebrate taxa and these differed in the weighting protocols used: differential weighting by codon position (all third positions or only third position transitions downweighted), by codon (codons representing labile residues downweighted), and combinations of both. Finally, amino acid changes were mapped unto the known phylogeny to find patterns of change corresponding to structural features of the protein.
The most accurate results of the phylogenetic tests were produced when labile codons were differentially weighted and third position transitions filtered from the analysis.
One conclusion from this study is that silent and non-synonymous sites should be evaluated and analyzed differently. Differential weighting by nucleotide position and type of change is effective for filtering noise at silent sites, i.e. filtering third position transitions but retaining third position transversions. Differential weighting by nucleotide position is not appropriate for replacement changes; mutations may be correlated among the three positions. Downweighting codons representing labile residues is effective in alleviating problems in retrieving phylogenetic signal at non-synonymous sites.
All of these studies have found that protein structure and function effects rates of gene evolution. Naylor and Brown (1997) found that the chemical properties, charge, and hydrophobicity of amino acids had significant effects on phylogenetic informativeness. Griffiths (1998) found that variation is correlated with specific locations in secondary structures, and that variation is constrained by interactions between residues important for secondary and tertiary structure and by interactions between nucleotides within codons. Goldman et al. (1998) found differences in rates of change related to secondary structure: rates for buried helices and sheets were greater than for buried turns and coils, and rates for exposed helices greater than those for other exposed residues. Koshi and Goldstein (1998) found that a model of evolution using parameters that incorporated location specific site heterogeneity produced improved the results of phylogenetic inference.
The assumption made when using data in phylogenetic analyses is that the data reflect the history of the taxa used. If variations in the data reflect processes other than history, recovering the historical signal may be compromised. If variation reflects constraints imposed by the requirements of the structure or function of a protein product, then adjusting the model to accomodate these processes is a necessary step in retrieving accurate phylogenetic hypotheses.
ACKNOWLEDGMENTS
This work would not have been possible without valuable discussions with G. Barrowclough, J. Bates, K. Burns, N. Caithness, J. Groth and S. Hackett and N. Klein and R. Rockwell. This work was supported by a grant from the Frank M. Chapman Memorial Fund of the American Museum of Natural History. The research reported in this paper is a contribution from the Lewis B. and Dorothy Cullman Research Facility at the American Museum of Natural History and has received generous support from the Lewis B. and Dorothy Cullman Program for Molecular Systematics Studies, a joint initiative of the New York Botanical Garden and the American Museum of Natural History.
REFERENCES
Brower, A. & Desalle, R. 1994. Practical and theoretical considerations for choice of a DNA sequence region in insect molecular systematics, with a short review of published studies using nuclear gene regions. Annals of the Entomological Society of America 87:702-716.
Brown, W.M. 1983. Evolution of animal mitochondrial DNA. In: Nei, M. and Koehn, R. K. (eds). Evolution of genes and proteins. Sunderland; Sinauer: 62-88.
Brown, W.M. 1985. The mitochondrial genome of animals. In: MacIntyre, R. J. (ed.). Molecular evolutionary genetics. New York; Plenum Publishing Corporation: 95-130.
Brown, W.M., George, M., Jr., & Wilson, A.C. 1979. Rapid evolution of animal mitochondrial DNA. Proceedings of the National Academy of Sciences 76: 1967-1971.
Brown, W.M., Prager, E.M., Wang, A. and Wilson, A.C. 1982. Mitochondrial DNA sequences of primates: tempo and mode of evolution. Journal of Molecular Evolution 18: 239-255.
Degli Esposti, M., De Vries, S., Crimi, M., Ghelli, A., Patarnello, T. and Meyer, A. 1993. Mitochondrial cytochrome b: evolution and structure of the protein. Biochimica et Biophysica Acta 1143: 243-271.
Edwards, S. V., Arctander, P. & Wilson, A.C. 1991. Mitochondrial resolution of a deep branch in the genealogical tree for perching birds. Proceedings of the Royal Society of London B 243:99-107.
Friesen, V.L. & Anderson, D.J. 1997. Phylogeny and evolution of the Sulidae (Aves: Pelecaniformes): A test of alternative modes of speciation. Molecular Phylogenetics and Evolution 7:252-259.
Goldman, N, Thorne, J.L. & Jones, D.T. 1998. Assessing the impact of secondary structure and solvent accessibility on protein evolution. Genetics 149:445-458.
Graybeal, A. 1993. The phylogenetic utility of cytochrome b: lessons from bufonid frogs. Molecular Phylogenetics and Evolution 2: 256-269.
Griffiths, C. 1997. Correlation of functional domains and rates of nucleotide substitution in cytochrome b. Molecular Phylogenetics and Evolution. 7: 352-365.
Griffiths, C. 1998. The correlation of protein structure and evolution of a protein-coding gene: Phylogenetic inference using cytochrome oxidase III. Molecular Biology and Evolution 15 (in press).
Hackett, S. 1996. Molecular phylogenetics and biogeography of tanagers in the genus Ramphocelus. Molecular Phylogenetics and Evolution. 5: 368-382.
Helm-Bychowski, K. & Cracraft, J. 1993. Recovering phylogenetic signal from DNA sequences: Relationships within the corvine assemblage (class: Aves) as inferred from complete sequences of the mitochondrial DNA cytochromeb gene. Molecular Biology and Evolution. 10: 1196-1214.
Howell, N. 1989. Evolutionary conservation of protein regions in the protonmotive cytochrome b and their possible roles in redox catalysis. Journal of Molecular Evolution 29: 157-169.
Irwin, D.M., Kocher, T.D. & Wilson, A.C. 1991. Evolution of the cytochrome b gene of mammals. Journal of Molecular Evolution. 32: 128-144.
Kim, J. 1996. General inconsistency conditions for maximum parsimony: effects of branch lengths and increasing numbers of taxa. Systematic Biology 45:363-374.
Kornegay, J.R., Kocher, T.D., Williams, L.A. & Wilson, A.C. 1993. Pathways of lysozyme evolution inferred from the sequences of cytochrome b in birds. Journal of Molecular Evolution. 37: 367-379.
Koshi, J.M. & Goldstein, R.A. 1998. Models of natural mutations including site heterogeneity. Proteins 28: (In press).
Moritz, C., Dowling, T.E. & Brown, W.M.. 1987. Evolution of animal mitochondrial DNA: relevance for population biology and systematics. Annual Review of Ecology and Systematics 18: 269-292.
Naylor, G.J.P & Brown, W.M. 1997. Structural biology and phylogenetic estimation. Nature 388, 528-530.
Olmstead, R.G. & Sweere, J.A. 1994. Combining data in phylogenetic systematics: an empirical approach using three molecular data sets in the solanaceae. Systematic Biology 43:467-481.
Sibley, C.G. & Ahlquist, J.E. 1987. Avian phylogeny reconstructed from comparisons of the genetic material, DNA. In: Patterson, C. (ed.). Molecules and morphology in evolution: Conflict or compromise? Cambridge; Cambridge University Press: 95-121
Simon C., Frati, F., Beckenback, A., Crespi, B., Liu, H. & Flook, P. 1994. Evolution, weighting, and phylogenetic utility of mitochondrial gene sequences and a compilation of conserved polymerase chain reaction primers. Annals of the Entomological Society of America 87: 651-698.
Sullivan, J. 1996. Combining data with different distributions of among-site rate variation. Systematic Biology 45:375-378.
Swofford, D.L., Olsen, G.L., Waddell, P.J. & Hillis, D.M. 1996. Phylogenetic inference. In: Hillis, D. M. , Moritz, C. and Mable, B. K. (eds.). Molecular systematics. 2nd ed.; Sunderland; Sinauer: 407-514.
Tsukihara, T., Aoyama, H., Yamashita, E., Tomizaki, T., Yamaguchi, H., Shinzawa-itoh, K., Nakashima, R., Yaono, R. & Yoshikawa, S. 1996. The whole structure of the 13-subunit oxidized cytochrome c oxidase at 2.8
a. Science 272 :1136-1144.Yoder, A.D., Vilgalys, R. & Ruvolo, M. 1996. Molecular evolutionary dynamics of cytochrome b in strepsirrhine primates: The phylogenetic significance of third-position transversions. Molecular Biology and Evolution 13: 1339-1350.
Table 1. Saturation assessments of subsets of cytochrome b sequences partitioned into functional regions.
Fig 1. Graphs illustrating the theoretical expectation of saturated and unsaturated data when sequence differences between pairs of taxa are plotted against time since divergence of those taxa. (A) Substitutions between taxa are not saturated. Pairwise differences increase over time. (B) Substitutions saturate. Pairwise differences reach a maximum, and the curve asymptotes at the theoretical maximum level of divergence.