S13.3: Nuclear gene introns versus mitochondrial genes as molecular clocks
William S. Moore1, Sam M. Smith2 & Thomas Prychitko1
1Department of Biological Sciences, Wayne State University, Detroit, Michigan, USA, e-mail
wmoore@biology.biosci.wayne.edu fax 313 577 6891; 2Clover Technologies, Inc., 1 Clover Court, Wixom, Michigan, USA 48393, USA, e-mail ssmith@clover.com Moore, W.S., Smith, S.M. & Prychitko, T. 1999. Nuclear gene introns versus mitochondrial genes as molecular clocks. In: Adams, N.J. & Slotow, R.H. (eds) Proc. 22 Int. Ornithol. Congr., Durban: 745-753. Johannesburg: BirdLife South Africa.A good molecular clock would be a powerful tool for studying the tempos and processes of evolution. Most evolutionary biologists would agree that there is no global molecular clock but local molecular clocks, calibrated from and applied to relatively closely related species, work reasonably well. With the objective of improving molecular clock analyses we developed a computer simulation model that enabled us to compare the clock properties of two alternative genes, mitochondrial encoded cytochrome b (cyt b) and nuclear encoded b -fibrinogen intron 7 (b fibint7). Simulations of the evolution of these genes indicate that b fibint7 should be an accurate clock for divergences ranging from recent to as old as 60 MY. For divergences less than approximately 5 MYA, cyt b is a better clock because its sampling variance is less than that of b fibint7, but it would dramatically underestimate older dates. The application of the two molecular clocks to woodpecker evolution suggests that the lineage leading to the subfamily Picinae (woodpeckers) separated from its sister lineage leading to the family Picumninae (piculets) approximately 15 MYA, that the radiation of the modern woodpeckers began about 7-8 MYA and that divergences between North American and South American taxa are more recent.
INTRODUCTION
It would be difficult to imagine a tool more powerful than a molecular clock for studying evolutionary tempos and processes. Even a somewhat imprecise clock would allow testing alternative hypotheses about the relationship of evolutionary history to specific geological or climatic events or about the rate of evolution both with regard to anatomical change and diversification of species. Klicka and Zink (1997), for example, argued that the average genetic distance between sister species of North American oscine birds is roughly 10 times greater than expected if many North American species arose in the late Pleistocene, as was widely thought among ornithologists. Even if the molecular clock applied by Klicka and Zink were somewhat imprecise, this order of magnitude disparity forces a radical re-thinking of the evolutionary history of the North American fauna and serious consideration of the hypothesis that many ‘ modern’ species arose considerably earlier in the Pliocene.
Since its inception (Zuckerkandl and Pauling, 1965), the molecular clock hypothesis has been odd among scientific hypotheses in that it has gained neither a broad consensus of support nor has it been clearly refuted. At this juncture, it is clear that there is no global molecular clock (i.e., a clock that works for all taxa across all expanses of time), but it is equally clear that rates of nucleotide substitution (mutations that become fixed in an evolutionary lineage), particularly of non-coding nucleotides, are sufficiently constant within groups of fairly closely related species to allow application of a ‘ local’ molecular clock. The contention over the molecular clock stems from the very nature of the clock. The clock is based on mutations, which are random events, but also, facets of a species biochemistry and natural history affect its mutation rates. These include DNA repair mechanisms, metabolic rate, and generation time. Much of the work to date on molecular clocks, and the contention over molecular clocks, has focused on testing rate constancy and on biological factors that might affect rates, and relatively little work has focused on the stochastic nature of the clock, problems this poses and solutions to those problems.
In this paper, we explore a few of the many stochastic aspects of molecular clocks. We do so from the perspective of accepting the molecular clock, at least among closely related species, as reasonably well established, but at the same time we acknowledge that it is not a very good timepiece. The question is: How best to make-do with an imprecise timepiece? Specifically, we present results from a computer model we developed that simulates the evolution of DNA sequences. We apply the model to simulate the sampling distributions of clock times (branch lengths) for two genes that have proved useful in avian phylogenetic analysis, the mitochondrial encoded cytochrome b gene (cyt b), a protein coding gene, and b -fibrinogen intron 7 (b fibint7), an intron of a nuclear encoded gene. DNA sequences used to estimate parameters required by the simulation model and the evolutionary time estimates derived from the model are those of woodpeckers.
Inasmuch as the theory of the molecular clock is based on adaptively neutral mutations, it is logical to choose DNA sequences for a molecular clock study that are adaptively neutral (introns) or to focus on neutral categories of nucleotides in functional sequences (synonymous nucleotides in protein coding genes). Although there is evidence that some nucleotides in some introns function in regulating gene expression, most nucleotides appear to be adaptively neutral in introns. Nucleotide substitutions in b fibint7 over the evolution of woodpeckers appear to be random, suggesting adaptive neutrality (Prychitko and Moore, 1997). Extending this logic to cyt b, we assembled a set of ‘clock’ sequences for woodpeckers that include only the 4-fold synonymous (4-fs) sites; i.e. third positions in codons where any of the four nucleotides specify the same amino acid. Although most substitutions involving 2-fold synonymous sites would be neutral, as doubtless are some nonsynonymous substitutions, the 4-fs set is the surest bet as a set of nucleotides that satisfy the assumptions of clock theory. Using the computer model, we compare the clock properties of these two DNA sequences over the evolution of woodpeckers and extrapolate from this analysis which gene should be the best molecular clock. It is apparent from the simulations that b fibint7 is expected to be a much more accurate molecular clock than cyt b for all but the most recent 4 to 5 MY of evolution. However, cyt b is expected to be a better clock for this most recent period because it is as accurate as b fibint7, but the variance of its sampling distribution is less than that of b fibint7.
METHODS
Published DNA sequences were used to estimate model parameters and to serve as ancestral sequences: cytochrome b (Moore and DeFilippis, 1997); b -fibrinogen intron 7 (Prychitko, 1997). The 4-fs sites sequences for cyt b (cyt b 4-fs) were constructed from the cyt b sequences using MEGA (Kumar, et al., 1993). The simulation program was written in Visual Basic (S. Smith) and is available at: http://www.science.wayne.edu/~biology/wsuevolution/mitomixer.html.
The computer model is a reiterative model that simulates DNA sequences evolving from a common ancestral sequence. Each iteration represents a period of time (1000 years in this study) during which each nucleotide (A, C, G, T) in the sequence can remain the same or be substituted by one of the other three nucleotides; the probabilities of these events are determined by a substitution probability matrix. The substitution matrices (Table 1) were estimated from nucleotide sequences of cyt b 4-fs and b fibint7 of woodpeckers by maximum likelihood using PAUP* (test version 4.0d64 written by D.L. Swofford). Estimation of the substitution matrix begins with estimation of the R-matrix, which is derived from the Q-matrix of the General Reverse Markov Process model (Yang, 1994; D. Swofford, personal communication). The columns and rows of the 4 x 4 R-matrix correspond to the nucleotides (A, C, G, T; Table 1), and the elements of the R-matrix are estimated ‘rate ratios’ that are proportional to rates of substitutions over the evolutionary history of the set of aligned sequences (Yang, 1994); e.g., RCT is proportional to the rate of C ® T substitutions. The Q- and R-matrices are related by Qij = Rij
p j, where p j is the equilibrium frequency of the target nucleotide (i.e. the nucleotide that replaces the previous nucleotide) and Qij and Rij are the elements in the ith row and jth column of the respective matrices. Knowledge of the Q-matrix is not important in this study and will not be discussed further. The substitution matrices (top value in each cell) and the R-matrices (parenthetical value in each cell) are presented in Table 1 for cyt b and b fibint7. The observed nucleotide frequencies, estimates of the p j’s, are given parenthetically with the column headings.The R-matrix can be transformed to an appropriate substitution matrix by scaling in accordance with a molecular clock calibration. An exact calibration is not critical for this study; so, we used the substitution rate of 2% /MY between two cyt b genes diverging from an ancestral sequence, or 1% along a single branch. Although this estimate is viewed as an approximation, it probably is not far off the actual value. Several molecular clock calibrations have been reported for avian species and all are consistent with a rate of divergence between species of approximately 2%/ MY (reviewed by Klicka and Zink, 1997). In addition, W.S. Moore derived an estimate for woodpeckers based on a fairly accurately dated skull (Becker, 1986; 2.9-3.5 MY, G.R. Smith, personal communication) that unequivocally belongs to the woodpecker genus Colaptes (Becker, 1986; W.S. Moore unpublished data and analysis); this analysis gave a cyt b substitution rate estimate of 2.0% ± 0.5% /MY, assuming the fossil was accurately dated at 3.2 MY. The rate of 2% /MY is a rate based on all sites of the cyt b gene. We transformed this value to a rate for cyt b 4-fs only. To do this we calculated the proportion of all substitutions that were 4-fs for each of two pairs of closely related species, Colaptes auratus vs. Piculus rubiginosus and Veniliornis callonotus vs. V. nigriceps. The species in each pair are sufficiently closely related that bias resulting from multiple hits should be negligible. For the two comparisons, 42 of 78 and 19 of 38 substitutions were 4-fs, respectively; thus, we estimate the proportion of 4-fs substitutions to be 0.53. This is a crude estimate and should be considered no more than a first approximation. Our cyt b sequences average 1047 nucleotides in length; thus, if the substitution rate is 1%/ MY along a branch, we expect 0.01 x 1047 = 10.47 substitutions/ MY, of which, 0.53 x 10.47 = 5.55 are expected to be 4-fs. Of the 1047 sites, 210 are 4-fs giving a substitution rate of 5.55/ 210 = 0.0264 substitutions/ 4-fs site/ MY (or .0000264/ 1000-year cycle).
The R-matrix values were transformed to conditional probabilities of substitution, according to the following procedure, as illustrated for cyt b 4-fs sites (Table 1): The diagonal elements represent probabilities that the nucleotide does not change during a 1000-year cycle; i.e., 1 - .0000264 = .9999736. The three remaining elements in each row of the R-matrix are normalised by summing the three R values, dividing each R-value by this sum and multiplying the quotient by 2.64x10-5. The result is that the four conditional probabilities represented in each row sum to one. For example, P(A® C|A) is (.024902657/ 4.1806914) x 2.64x10-5 = 1.57254x10-7. Verbally, this is the conditional probability of the substitution of an A by a C given that that the nucleotide is an A at the beginning a 1000-year reiteration cycle.
The b fibint7 substitution matrix (Table 1) was derived analogously but with a molecular clock calibration of 2.53 x 10-3 substitutions/ nucleotide/ MY (or 2.53 x 10-6/ 1000-year cycle), which reflects the lower substitution rate of b fibint7 relative to 4-fs cyt b sites. The clock calibration for b fibint7 was determined by regression through the origin of genetic distances for b fibint7 on cyt b for pairs of woodpecker species for which both sequences were available. There were 92 distance pairs, but it is apparent from inspection of the plot (not shown) of b fibint7 distances against cyt b 4-fs distances that the relationship becomes non-linear at high distance values of cyt b 4-fs as a result of multiple substitutions. Thus, the regression coefficient was estimated from 70 data points over the range of values for which the relationship appears linear. The regression coefficient is 0.096 b fibint7 sub./ cyt b 4-fs. sub.; thus, (0.096) x (2.64 x 10-5) subs/ nuc./ 1000-year cycle = 2.53 x 10-6 subs/ nuc./ 1000-year cycle.
The substitution matrices ‘drive’ the simulated evolution of the cyt b 4-fs and b fibint7 sequences. In each iteration, beginning with the ancestral sequences, each nucleotide is subjected to a multinomial trial with outcomes (stay the same or change to one of the other three nucleotides) determined by the conditional probabilities specified in the appropriate row of the substitution matrix. At the end of the cycle, the ‘evolved’ sequences become the ancestral sequences for the next iteration. Evolution is simulated by reiterating this process; e.g., 20000 iterations simulates 20 MY of sequence evolution. The model is ‘primitive’ in the sense that it simulates evolution as a series of multinomial trials rather than using the Poisson, negative binomial or normal distributions as approximations of the more complex (and unknown) underlying distributions. This has the advantage of providing a more accurate simulation of the complex, underlying, stochastic phenomenon, but the program requires considerable computation time.
Because the substitution matrices were estimated from real data and it is a primitive model, the simulations should closely approximate the real evolution of the cyt b 4-fs and b fibint7 sequences for woodpeckers. Presently the simulation program is considered a b -test version, but we have extensively, if not exhaustively tested it. Details of the simulation model will be published elsewhere; suffice it to say that when the simulations are run for a specified number of cycles corresponding to the estimated time of divergence between woodpecker species, the program returns simulated distributions of genetic distances and clock times in agreement with expectations.
RESULTS
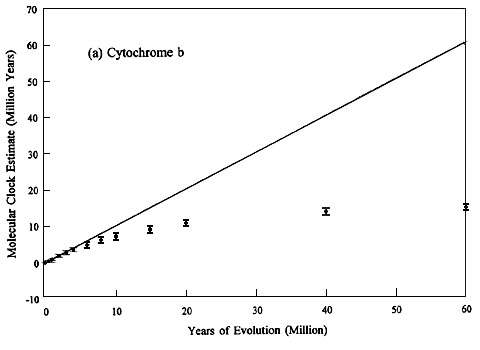
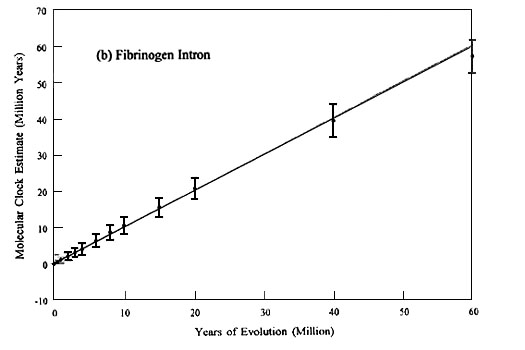
We simulated 100 replicates of cyt b 4-fs and b fibint7 evolution for time spans ranging from 0.5 to 60 MY. The results of the cyt b 4-fs simulations are summarised in Fig. 1A and those of b fibint7 in Fig. 1B. Each replicate was started with the same Colaptes auratus (Northern Flicker) cyt b 4-fs and b fibint7 sequences, and 1000-year periods were reiterated to completion of the specified time (horizontal axis). The resultant distributions represent clock times that would result if one were able to replicate repeatedly the evolution of these genes. The dots indicate the means of the simulated distributions and the vertical bars indicate ± 1 S.D. The solid line represents the trace of a perfect molecular clock; i.e., the clock estimate is identical to the actual years of evolutionary history.
DISCUSSION
The most striking and practical conclusion that can be drawn from this study is that b fibint7 should be a relatively good molecular clock for divergences up to, certainly, 40 MY and perhaps as much as 60 MY; whereas, the accuracy of the cyt b 4-fs clock diminishes rapidly after about five MY. However, although the means of the b fibint7 and cyt b 4-fs distributions are both close to the ideal molecular clock for the most recent five MY of evolution, cyt b 4-fs is the better molecular clock over this time span because the variance of its sampling distribution is less than that of b fibint7. In other words, both have similar accuracy but the precision of the cyt b 4-fs clock is better. Beyond approximately five MY the cyt b 4-fs clock becomes so inaccurate that precision doesn’t matter, and b fibint7 is clearly the better molecular clock.
It is useful in thinking about ways to
improve molecular clocks to examine the underlying probability distribution and what is
being estimated. Molecular clock estimates are based on statistical samples of nucleotide
sites. The number of accrued substitutions between two gene sequences that have diverged a
period of time T can be modelled by a binomial distribution, where n is the
number of sites (trials) and p is the probability that a given site is substituted
(success). The usual assumptions of binomial models apply; viz., sites are independent and
p is constant over sites. These are reasonably satisfied for neutral mutations when
divergence is low. If it has been established that divergence is clock-like, then p = l T, where l corresponds to the rate
parameter of the molecular clock and is usually estimated from the fossil record in a
separate analysis; p is the binomial probability of a substitution and is estimated
from the data as ![]() (x
= number of substitutions in a sequence of length n). The ages of unknown
divergences (T) in the phylogeny are then estimated from the relationship
(x
= number of substitutions in a sequence of length n). The ages of unknown
divergences (T) in the phylogeny are then estimated from the relationship ![]() .
. ![]() has a binomial distribution because it is the
number of substitutions divided by a constant; i.e.,
has a binomial distribution because it is the
number of substitutions divided by a constant; i.e., ![]() . As is characteristic of binomial distributions,
the variance of the sampling distribution of T is diminished as the number of
trials (number of nucleotides) increases. Thus, increasing the number of nucleotides
sampled (i.e., the length of the sequence) should improve the precision of the
clock estimate. In the case of cyt b, most of the substitutions are synonymous and it is
likely that synonymous sites at the other 12 protein coding genes in the mitochondrial
genome are substituted at the same rate. Thus, sequence from several protein coding genes
could contribute to a large pool of sites for estimating the rate parameter. Similarly,
nuclear genes usually have several introns. b -fibrinogen has
seven, which opens the possibility that as many as seven closely linked sequences could be
pooled to create, in effect, a larger number of trials and a correspondingly smaller
variance of the estimate of the rate parameter. To explore the effect of increasing the
size of the nucleotide pool on the precision of the clock estimate, we did a series of
simulations with the length of the sequences doubled by simply ‘ duplicating’
each gene. Thus, the information content is the same, but the number of nucleotide sites
is doubled. The means and standard deviations for simulated molecular clock estimates for
several time expanses are summarised in Table 2. As expected, the
standard deviation of the sampling distribution decreases for the doubled number of
nucleotides, but the standard deviation is not halved. Although further increasing the
number of nucleotides yields a smaller standard deviation, there is a diminishing return.
. As is characteristic of binomial distributions,
the variance of the sampling distribution of T is diminished as the number of
trials (number of nucleotides) increases. Thus, increasing the number of nucleotides
sampled (i.e., the length of the sequence) should improve the precision of the
clock estimate. In the case of cyt b, most of the substitutions are synonymous and it is
likely that synonymous sites at the other 12 protein coding genes in the mitochondrial
genome are substituted at the same rate. Thus, sequence from several protein coding genes
could contribute to a large pool of sites for estimating the rate parameter. Similarly,
nuclear genes usually have several introns. b -fibrinogen has
seven, which opens the possibility that as many as seven closely linked sequences could be
pooled to create, in effect, a larger number of trials and a correspondingly smaller
variance of the estimate of the rate parameter. To explore the effect of increasing the
size of the nucleotide pool on the precision of the clock estimate, we did a series of
simulations with the length of the sequences doubled by simply ‘ duplicating’
each gene. Thus, the information content is the same, but the number of nucleotide sites
is doubled. The means and standard deviations for simulated molecular clock estimates for
several time expanses are summarised in Table 2. As expected, the
standard deviation of the sampling distribution decreases for the doubled number of
nucleotides, but the standard deviation is not halved. Although further increasing the
number of nucleotides yields a smaller standard deviation, there is a diminishing return.
As an application of some of the ideas in this paper, we have estimated divergence times for a few woodpecker lineages (Table 3). These divergence estimates are, of course, only first approximations because our calibration of the molecular clock with woodpecker fossils needs to be extended and refined; nonetheless, they provide a coarse-focus overview of the chronology of woodpecker evolution that should not be grossly inaccurate. The clock estimates are based on the p-distance between species (or averages of several species in a clade) representing distinct lineages: for cyt b 4-fs sites, the divergence rate between 2 lineages was taken to be 2 x the rate along a single branch (see above) or 2 x (2.64 x 10–2) subs./ nuc./ MY = 5.28 x 10–2 subs./ nuc./ MY and 2 x (2.53 x 10-3) subs/ nuc./ MY = 5.06 x 10–3subs/ nuc./ MY for b fibint7. The estimated divergence times for cyt b 4-fs and b fibint7 presented in Table 3 were calculated by dividing the observed (or averaged) p-distances between the taxa indicated by 5.28 x 10–2 and 5.06 x 10–3, respectively.
The youngest divergence is that of the South American genus Veniliornis from the North American species Picoides villosus. Estimated to be 3.98 MYA by cyt b and 3.43 MYA by b fibint7. Based on our simulations, the older age provided by the cyt b 4-fs clock is probably closer to the actual value. The divergence of Colaptes auratus and C. rupicola + Piculus rubiginosus represents the split between the North and South American Flickers. (P. rubiginosus is presently incorrectly classified and should be assigned to the genus Colaptes, Moore and DeFilippis, 1997). Here, again, the cyt b 4-fs is probably the closer value(4.57 MY). The most unexpected estimates are for the divergence of the sapsuckers (Sphyrapicus) from the melanerpine woodpeckers. This occurred at a time depth when cyt b would be expected to underestimate the actual age, but in this case cyt b gives the older estimate, 7.51 MYA, versus 5.04 MYA for b fibint7. It is reasonable to infer that the sapsuckers and melanerpines shared a common ancestor 5.0 – 7.5 MYA. The estimated age for the base of the Picinae (modern woodpeckers) was made by comparing the average p-distance from Sphyrapicus and Melanerpes to all other woodpeckers; this divergence appears to be close to the base of the tree (Moore and DeFilippis, 1997; Prychitko and Moore, 1997). At this time depth, the age estimated from the b fibint7 clock, 7.41 MYA, should be more accurate. Finally, the separation of the true woodpeckers (Picinae lineage) from the piculets (Picumnus aurifrons) dates to 15.06 MYA by the b fibint7 clock versus 8.74 by the cyt b 4-fs clock. Our simulations suggest that the b fibint7 date is much more accurate. These clock estimates clearly would be improved by obtaining additional sequence from other mitochondrial-encoded protein genes and other b -fibrinogen introns.
ACKNOWLEDGEMENTS
We thank Shannon Hackett and John Bates for inviting us to participate in this symposium and David Webb for critically reading an early draft of the manuscript. This work was supported in part by grants from the Systematic Biology Program, National Science Foundation, USA, DEB-9316452 and DEB-9726512 to WSM.
REFERENCES
Becker, J.J. 1986. Fossil birds of the Oreana local fauna (Blancan), Owyhee County, Idaho. Great Basin Naturalist 46: 763-768.
Klicka, J. & Zink, R.M. 1997. The importance of recent ice ages in speciation: a failed paradigm. Science 277: 1666-1669.
Kumar, S., Tamura, K. & Nei, M. 1993. Mega: Molecular Evolutionary Genetics Analysis, version 1.01, University Park.
Moore, W.S. & Defilippis, V.R. 1997. The window of taxonomic resolution for avian phylogenies based on mitochondrial cytochrome b DNA sequences. In: D. P. Mindell (ed.) Molecular evolution and systematics; New York; Academic Press: 83-119..
Prychitko, T. 1997. Utility of an intron from the B-fibrinogen gene in phylogenetic analysis of woodpeckers (Aves: Picidae). Doctoral thesis, Wayne State University.
Prychitko, T.M. & Moore, W.S. 1997. The utility of DNA sequences of an intron from the beta-fibrinogen gene in phylogenetic analysis of woodpeckers (Aves: Picidae). Molecular Phylogenetics Evolution. 8: 193-204.
Tavare, S. 1986. Some probabilistic and statistical problems on the analysis of DNA sequences. Lectures in mathematics in the life sciences 17: 57-86.
Yang, Z. 1994. Estimating the pattern of nucleotide substitution. Journal of Molecular Evolution. 39: 105-111.
Zuckerkandl, E. & Pauling, L. 1965. Evolutionary divergence and convergence in proteins. In: Bryson, V. and Vogel, H. J. (eds.) Evolving genes and proteins; New York; Academic Press: 97-165.
Table 1. Substitution probability matrices and R-matrices for woodpecker DNA sequences, based on maximum likelihood estimation using the General Reversible Markov (Tavare, 1986; Yang, 1994; PAUP*).
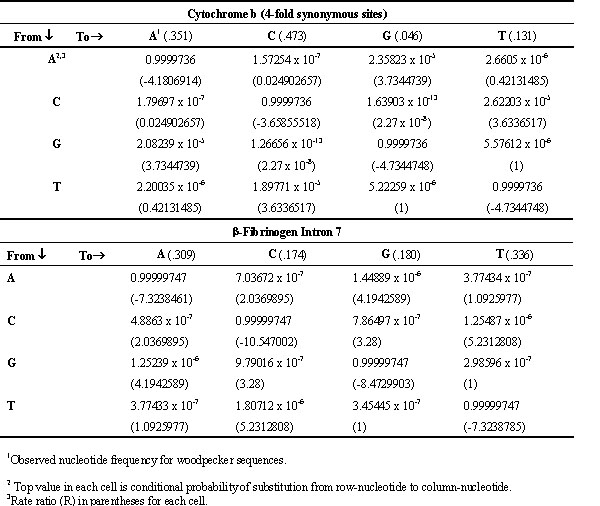
Table 2. Decrease in standard deviation of molecular clock estimates as a result of doubling the length of DNA sequence. Values in the body of the table are means of simulated molecular clock estimates in millions of years ± 1 S.D.
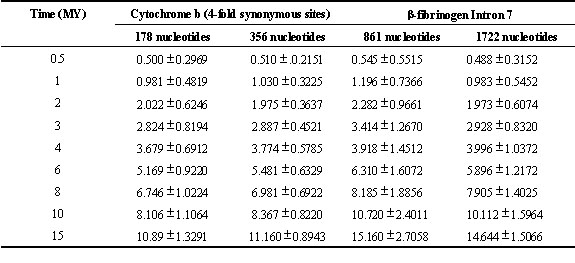
Table 3. Molecular clock estimates of divergence times for some woodpecker lineages.
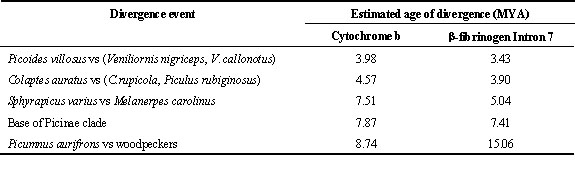
Fig. 1. Molecular clock estimates of evolutionary time for (a) cytochrome b 4-fold synonymous sites and (b) b fibrinogen intron 7 plotted against actual (simulated) years of evolution. Dots are means of 100 simulated replicates, the vertical bars are ± 1 S.D. The continuous line represents a perfect molecular clock, which gives an estimated time identical to the actual years of evolution.